

Repli-FISH (Fluorescence in Situ Hybridization): Application of 3D-(Immuno)-FISH for the Study of DNA Replication Timing of Genetic Repeat Elements
Patrick Weber † ![]() , Cathia Rausch †
, Cathia Rausch † ![]() , Annina Scholl
, Annina Scholl ![]() , M. Cristina Cardoso *
, M. Cristina Cardoso * ![]()
† These authors contributed equally to this work.
- Cell Biology and Epigenetics, Department of Biology, Technische Universität Darmstadt, 64287 Darmstadt, Germany
* Correspondence: M. Cristina Cardoso ![]()
OBM Genetics 2019, Volume 3, Issue 1 doi: 10.21926/obm.genet.1901062
Academic Editor: Thomas Liehr
Special Issue: Applications of Fluorescence in Situ Hybridization
Received: July 22, 2018 | Accepted: January 8, 2019 | Published: January 25, 2019
Recommended citation: Weber P, Rausch C, Scholl A, Cardoso MC. Repli-FISH (Fluorescence in Situ Hybridization): Application of 3D-(Immuno)-FISH for the Study of DNA Replication Timing of Genetic Repeat Elements. OBM Genetics 2019; 3(1): 062; doi:10.21926/obm.genet.1901062.
© 2019 by the authors. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
Abstract
Background: Genetic repeat elements (interspersed or tandem repeats) have diverse functions within cells and at different phases of the cell cycle. However, their investigation at a genome-wide scale is challenging due to their repetitive nature. Here, we describe a method to study the DNA replication kinetics of different repeat elements in single cells throughout the S-phase of the cell cycle. Methods: Mouse major satellite, minor satellite and telomere repeat elements as well as human LINE-1 and Alu repeats were detected in fluorescence in situ hybridization (FISH) assays using specific hybridization probes containing biotinylated, digoxigenin-conjugated or fluorescently-labeled nucleotides. We combined their visualization with the detection of cellular DNA replication signals at the single cell level. Results: Specific hybridization of all probes to their intended target sequence and their location on individual chromosomes were verified by microscopic analysis. DNA replication was detected by labeling replisome components or nascent DNA synthesis by detecting incorporated thymidine analogs. The different spatial distribution of replication signals allowed us to classify the cells in different S-phase substages. We also measured changes in genomic DNA content during the progression through S-phase. Using high-throughput/high-content microscopic analysis, we then performed colocalization analysis of the degree of overlap between DNA replication and DNA probe signals to determine the kinetics of DNA replication of repeat elements in individual cells. Conclusions: Application of these probes in 3-dimensionally preserved interphase nuclei with the simultaneous detection of newly synthesized DNA or components of the replication machinery allows determination of the replication kinetics of all elements during the synthesis phase of the cell cycle in human cells as well as in embryonic and somatic mouse cells.
Graphical abstract

Keywords
3D-FISH; Alu; DNA replication timing; EdU; LINE-1; PCNA; genetic repeat elements; satellite DNA; single cell analysis; telomeres
1. Introduction
DNA replication is a fundamental cellular process that, like DNA transcription and repair, requires tight control. Dysregulation of this process results in the introduction of mutations or chromosomal aberrations that lead to multiple disorders, including cancer. Thus, elucidation of the molecular mechanism responsible for the control of DNA replication dynamics is of great importance.
Many studies have shown that different chromatin types are replicated at distinct times during S-phase (reviewed in [1]). This is reflected by the changing spatial nuclear distribution of replication signals (visualized by thymidine analog incorporation or labeled replication machinery proteins) observed by light microscopy and termed replication patterns. Each pattern has been shown to correspond to the duplication of a different chromatin type, i.e., euchromatin, facultative and constitutive heterochromatin.
Recently established genome-wide replication timing protocols confirmed that the different chromatin types initiate DNA replication within specific zones (replication domains) along the genome [2]. It was further shown that a substantial proportion of the genome undergoes large-scale chromatin reorganization during embryonic development [2].
Repetitive DNA elements account for more than half of the human genome and up to 40% of the mouse genome (Figure 1C), whereas protein-coding sequences constitute only 1.2% and 1.4% of the human and mouse genomes, respectively [3,4]. In the human genome, repetitive and transposable genetic elements include, among others, LINEs (long interspersed nuclear elements) and SINEs (short interspersed nuclear elements). Both elements rely on reverse transcription of an RNA intermediate for integration into new genomic locations [5].
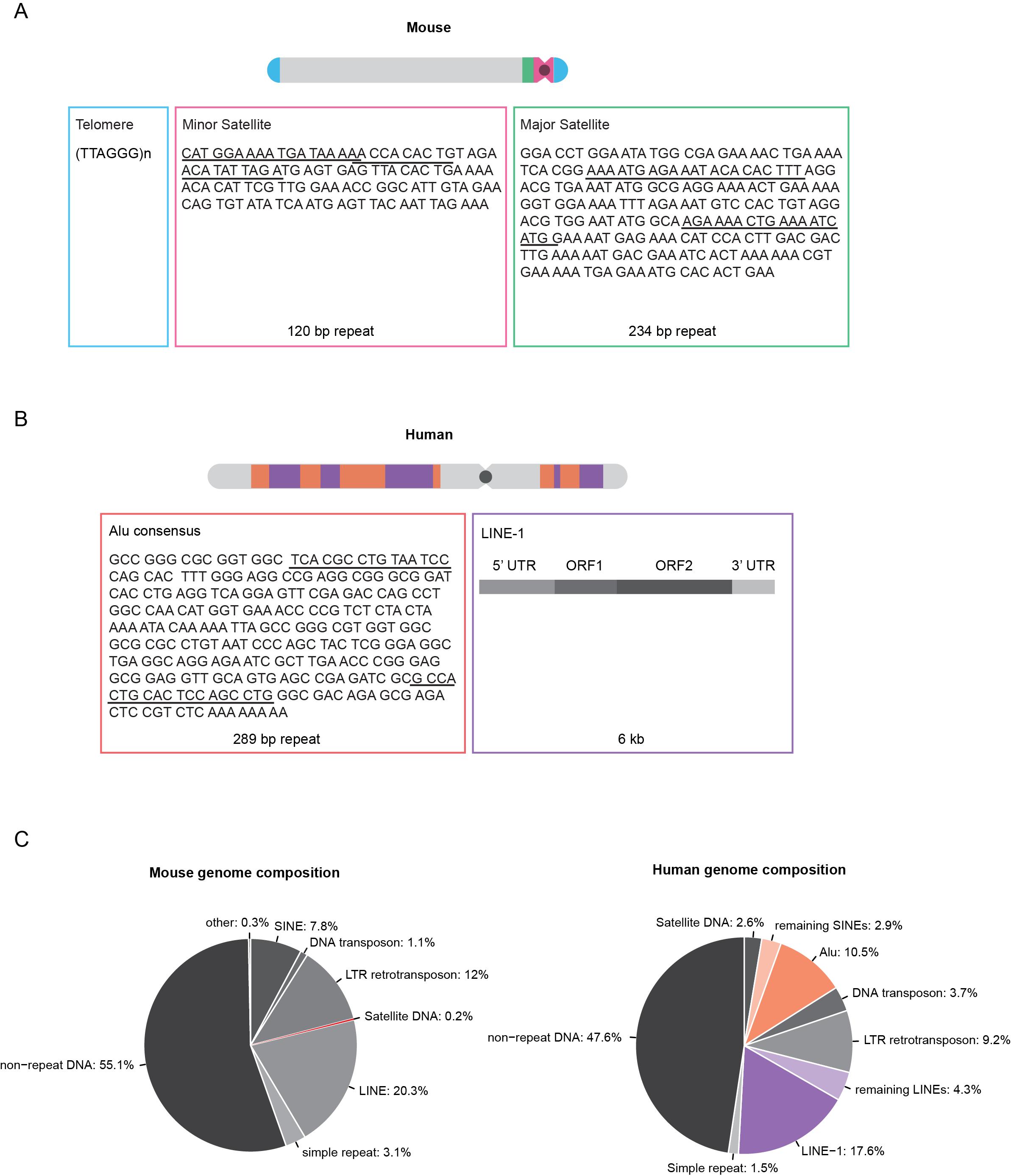
Figure 1 Overview of the localization and sequence of mouse and human repetitive elements. A: Schematic overview of an acrocentric mouse chromosome showing the localization of telomeric, minor and major satellite DNA. B: Schematic overview of a human chromosome showing the localization of Alu and LINE-1 elements. The sequence information for telomeres, minor and major satellite and Alu elements depict the consensus sequence of one repeat unit. Underlined sequence regions represent primer binding sites for PCR amplification of FISH probes. Full-length LINE-1 elements are 6 kb in length and consist of a 5’ and a 3’ untranslated region (UTR) and two open reading frames (ORF1 and ORF2). C: Pie charts showing the mouse (left) and human (right) genome composition (Repeat Masker). Approximately half of the genomes are composed of different classes of repetitive DNA elements.
LINE-1 elements represent a family of autonomous and active retroelements that account for approximately 17% of the human genome [3]. These elements are found in gene-poor and AT-rich regions, which correspond to dark G-bands on Giemsa-stained metaphase chromosomes [6]. The majority of LINE-1 elements are retrotransposition defective due to 5’ truncations, rearrangements or mutations, resulting in 80-100 retrotransposition active LINE-1 elements out of the half a million LINE-1 copies in the human genome [3,7]. Full-length LINE-1 elements have a total length of 6 kb and contain a 5’ untranslated region (UTR), three open reading frames (ORF) and a 3’ UTR ending in a AATAAA polyadenylation signal. ORF1 and ORF2 encode the p40 protein and a protein with endonuclease and reverse transcriptase activity that are necessary for retrotransposition [8,9].
Alu elements are part of the SINE family and represent the most abundant class of non-autonomous retrotransposons. Alu elements cover approximately 11% of the human genome and are predominantly found in gene-rich regions, which, in contrast to LINE-1 elements, correspond to dark R-bands on metaphase chromosomes stained by reverse-Giemsa banding [3,6]. Alu elements are approximately 290 bp long and have a dimeric structure, consisting of two highly similar left and right monomers separated by a linker sequence. The Alu element sequence does not encode proteins, although the left monomer contains a promoter for RNA polymerase III dependent transcription [3,10]. In trans retrotransposition is mediated by LINE-1-encoded proteins [11,12].
In mouse and human cells, telomeres are composed of non-coding, double-stranded TTAGGG array repeats bound by the shelterin complex [13,14]. The ends of linear chromosomes mirror DNA damage breaks; however, their repair would lead to chromosome fusions. Shelterin prevents telomere recognition, DNA damage responses and chromosome end processing via repair pathways. Similar to telomerase, shelterin also serves to regulate telomere length [14].
In the mouse genome, centromeric and pericentromeric DNA is composed of highly conserved, tandem repeats of DNA consisting of multiple copies of the repeat unit organized in a head-to-tail manner and referred to as minor satellites (MiSat) and major satellites (MaSat) according to their size. MiSats occupy approximately 600 kb of the terminal centromeric regions of all acrocentric mouse chromosomes and are composed of 120-bp AT-rich repeat units [15]. The pericentric MaSat DNA spans 6 Mb adjacent to the MiSat and consists of 234-bp repeats that form clusters in mouse cells, visible as bright structures following DAPI counterstaining and referred to as chromocenters [16].
Although historically referred to as “junk”, “parasitic” or “selfish” DNA due to the lack of a clear function within the genome, repetitive elements are now believed to play a role in genome organization during the cell cycle and throughout development as well as in disease conditions [17,18,19,20,21,22]. Complete and error-free DNA duplication prior to mitosis is an absolute requirement for the preservation of genome integrity and ultimately, the development and maintenance of an organism. This applies not only to the protein-encoding part of the genome, but also holds true for the repetitive elements that are interspersed throughout the genome [23]. However, due to their repetitive nature, repetitive elements are difficult to investigate in genome-wide studies and, hence, the spatio-temporal organization of the replication of repetitive DNA elements remains to be fully elucidated.
Here, we present a protocol that combines fluorescence in situ hybridization (FISH) using repeat-specific probes with the detection of DNA replication signals (Repli-FISH) and DNA content. This method can be applied to 3-dimensionally preserved cells and allows high-throughput microscopic analysis at the single cell level.
2. Materials and Methods
A timeline of a typical Repli-FISH experiment is outlined in Figure 2A.
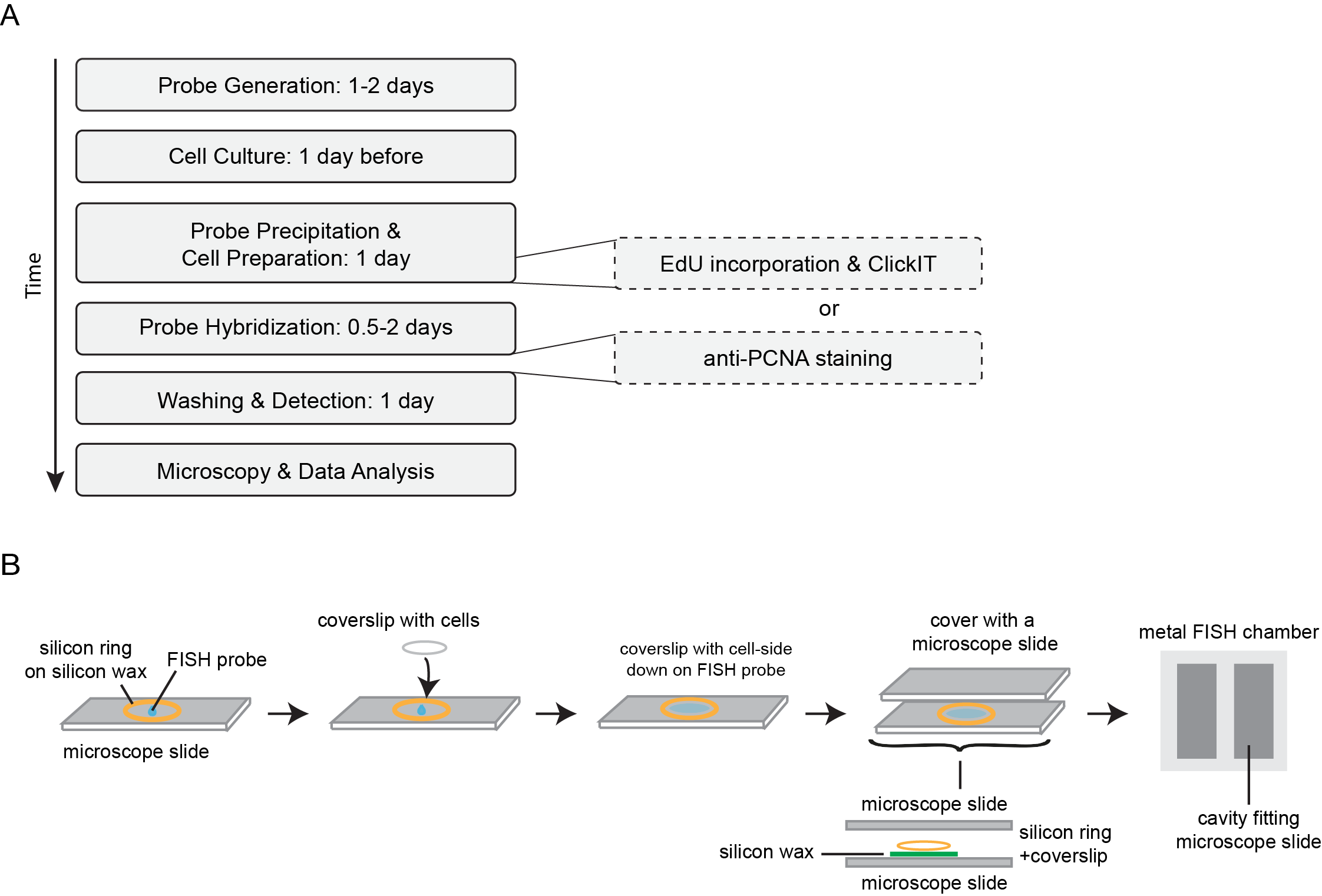
Figure 2 Representative timeline and setup of FISH experiments. A: Outline of the timelines of a typical replication FISH (Repli-FISH) experiment, showing that the combination of replication visualization and FISH can be performed over the course of four days. B: Experimental setup of the FISH denaturation and hybridization. A silicon wax mark is applied on a microscope slide and the coverslip is placed cell-side down on the probe; a silicon ring is used for additional sealing. A second microscope slide is then placed on top as a cover. For denaturation and hybridization, the entire apparatus is placed in a tightly sealed metal chamber.
2.1 Cell Culture
Human HeLa cervical carcinoma cells ([24], ATCC CCL-2) and mouse C2C12 myoblast cells ([25], ATCC CRL-1772) as well as mouse embryonic fibroblast cells (MEF W8, [26]) were cultured at 37°C under 5% CO2 in high glucose Dulbecco’s modified Eagle’s medium (DMEM; Cat. No.: D6429, Sigma–Aldrich Chemie GmbH, Steinheim, Germany) supplemented with 1 μM gentamicin (Cat. No.: G1397, Sigma–Aldrich Chemie GmbH), 20 mM L-glutamine (Cat. No.: G7513, Sigma–Aldrich Chemie GmbH) and fetal calf serum [FCS; 10% (HeLa and MEF W8) or 20% (C2C12)]. Cells were passaged every two days.
Murine embryonic stem cells (mESCs, J1, [27], ATCC SCRC-1010) were cultured in high glucose DMEM (Cat. No.: D6429, Sigma–Aldrich Chemie GmbH) containing 16% FCS, 1x non-essential amino acids (Cat. No.: M7145, Sigma–Aldrich Chemie GmbH), 1x penicillin/streptomycin (Cat. No.: P4333, Sigma–Aldrich Chemie GmbH), 1x L-glutamine (Cat. No.: G7513, Sigma–Aldrich Chemie GmbH), 0.1 mM beta-mercaptoethanol (Cat. No.: 4227, Carl Roth, Karlsruhe, Germany), PD 0325901 (0.1 µM; Cat. No.: Axon 1408, Axon Medchem BV, Groningen, The Netherlands), CHiR 99021 (0.3 µM; Cat. No.: Axon 1386, Axon Medchem BV) and LIF (1,000 U/ml; Cat. No.: ALX-201-242, Enzo Life Sciences GmbH, Lörrach, Germany). Cell culture medium was changed every day and cells were split every two days at a ratio of 1:8 – 1:10.
For Repli-FISH experiments, cells were seeded on gelatin-coated glass coverslips (0.2% gelatin; Cat. No.: G2500, Sigma–Aldrich Chemie GmbH).
2.2 Isolation of Genomic DNA
For the preparation of genomic DNA (gDNA), HeLa or C2C12 cells were harvested by centrifugation (10 min, 400 xg, 4°C) and incubated overnight at 50°C in TNES buffer (10 mM Tris [pH 7.5], 400 mM NaCl, 10 mM EDTA, 0.6% SDS and 1 mg/ml proteinase K (Carl Roth)). Subsequently, 0.6 mg/ml RNAse A (Qiagen, Hilden, Germany) was added and DNA was incubated for 30 min at 37°C to remove RNA. NaCl (6 M) was added to the extracted gDNA at a final concentration of 1.25 M. After centrifugation (15 min, 16,000 xg, room temperature), gDNA was precipitated from the supernatant by the addition of ice-cold 100% ethanol and incubation at -20°C for 1 h. The gDNA was pelleted by centrifugation (10 min, 16,000 xg, 4°C), washed with 70% ethanol, dried at room temperature and dissolved in ddH2O.
2.3 Bio-16-dUTP Nucleotide Generation
Bio-16-dUTP labeling reactions contained 4.5 mM aminoallyl-dUTPs (5-(-3-aminoallyl)-2’-deoxyuridine 5’-triphosphate, Cat. No: A 0410, Sigma–Aldrich Chemie GmbH), 44.5 mM carbonate-bicarbonate buffer (0.2 M NaHCO3 stock with 0.5 M NaCl titrated to pH 8.3 with 0.2 M Na2CO3) with 0.5 M NaCl and 8.9 mM biotin-XX (6-((6-((biotinoyl)amino)hexanoyl)amino)hexanoic acid, Cat. No: B-1606, Molecular Probes) in a total volume of 45 μl. Reactions were incubated for 3–4 hours at room temperature and stopped by the addition of 20 mM glycine (pH 8.0). Subsequently, 20 mM Tris-HCl (pH 7.75) was added to stabilize the nucleotides [28].
2.4 Probe Generation
Details of primers used for all PCR-based labeling reactions (except for the LINE-1 probe, which was generated by nick-translation) are shown in Table 1.
Table 1 Primers used for FISH probe generation.

2.4.1 Alu-Specific Probe
Alu-specific probes were generated via a two-step PCR. First, Alu elements were amplified from HeLa cell genomic DNA using partially degenerate Alu-specific primers (Table 1 and Figure 1A), followed by a PCR-based labeling with bio-16-dUTPs (Tables 2 and Tables 3). The product of the labeling PCR was subsequently purified with the QIAQuick PCR purification kit (Qiagen) according to the manufacturer’s instructions.
Table 2 Reaction setup for Alu-specific probe PCR.
Table 3 PCR cycling conditions for Alu-specific probe amplification.
2.4.2 LINE-1-Specific Probe
LINE-1 probes were generated and labeled with bio-16-dUTPs by nick-translation of the pLRE3-eGFP plasmid containing the LRE wild-type LINE-1 sequence ([33], a kind gift from John V. Moran, University of Michigan Medical School, USA). Reactions contained 50 mM Tris-HCl pH 8, 5 mM MgCl2, 0.5 mg/ml BSA, 10 mM beta-mercaptoethanol, 0.04 mM biotin-16-dUTPs or digoxigenin-11-dUTPs, 0.05 mM dATP/dGTP/dCTP each, 0.32 U DNase I (D5025, Sigma–Aldrich Chemie GmbH), 10 U Klenow fragment (M0210, NEB, Ipswich, MA, USA), 1 μg pLRE3-eGFP plasmid DNA in a total volume of 100 μL. Reactions were incubated for 90 min at 15°C and stopped by adding 5 μl 0.5 M EDTA.
Alu- and LINE-1-specific probes were sheared with a Covaris S220 device (Covaris Inc., Woburn, MA, USA) to a final size of approximately 250 bp in microTUBES (520045, Covaris Inc.). Quality and size were evaluated by 1% agarose gel electrophoresis.
2.4.3 Major and Minor Satellite Repeat-Specific Probes
Both major and minor satellite repeat-specific probes were generated via PCR amplification using repeat-specific primers (Table 1 and Figure 1) and mouse genomic DNA from C2C12 mouse myoblasts as the template (Tables 4 and Tables 5). Since this amplification results in probes of varying lengths (detectable as a smear following agarose gel electrophoresis, see Figure 3), samples need to be digested using DNase I (1:250 dilution in ddH2O (stock: 2U/µl), 2 µl/reaction) for a maximum of 30 min at room temperature. The reaction was stopped by the addition of 1 µl 0.5 M EDTA and probe quality and size were evaluated by 1% agarose gel electrophoresis (15 µl of reaction mixture).
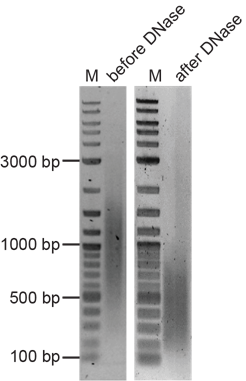
Figure 3 Assessment of FISH probe quality. Agarose gel showing probes before and after shearing. Amplification of repetitive DNA elements via PCR or nick-translation leads to amplicons of different sizes (left); digestion with DNase I or mechanical shearing decreases the length of the probes (right). M = marker.
Table 4 Reaction mixture for major and minor satellite-specific probe PCR amplification.
Table 5 PCR cycling conditions for major and minor satellite-specific probe amplification.
2.4.4 Telomere Repeat-Specific Probe
Probes specific for telomeric repeats were generated by a two-step PCR using previously described self-annealing primers [32]. The first PCR reaction served to create elongated probe fragments with regular dNTPs, while the second reaction included biotinylated or cyanine dye-conjugated nucleotide analogues for efficient probe labeling (Table 6). For the initial amplification, a high-fidelity polymerase was used (e.g. Q5 polymerase (NEB, Cat#: M0491)). For labeling with bio-16-, Cy3- or Cy5-conjugated dUTPs in the subsequent PCR, however, Taq polymerase was required since the Q5 polymerase does not incorporate dUTPs. Identical cycling conditions were used in both reactions (Table 7).
Table 6 Reaction mixture for telomere repeat-specific probe PCR amplification.
Table 7 PCR cycling conditions for telomere-specific probe amplification.
2.5 Probe Precipitation and Fluorescence in Situ Hybridization (FISH)
NOTE: Co-detection of incorporated EdU must be performed prior to FISH (see Figure 2A and refer to Section 2.6.2).
2.5.1 Probe Precipitation
Probes were ethanol-precipitated with 0.15 M sodium-acetate and 50 μg/ml fish sperm DNA, washed with 70% ethanol, air-dried and resuspended in hybridization solution containing formamide (70% formamide, 2x SSC (30 mM Na-citrate, 300 mM NaCl), 10% dextran sulfate, pH 7) for MaSat-, MiSat- and telomere-specific probes, and without formamide (10 mM Tris-HCl, 3 mM MgCl2, 50 mM KCl, 10 μg/ml gelatin, 2x SSC, [34,35]) for Alu- and LINE-1-specific probes. Probes were denatured for 5 min at 80°C and immediately placed on ice.
2.5.2 Cell Preparation
For subsequent hybridization and detection of replication signals, cells were washed twice with 1x PBS and immediately fixed with 3.7% formaldehyde/PBS for 10–15 min. After washing (2–3x) in PBS-T (0.02% Tween), cells were permeabilized in 0.5% Triton-X100/PBS for 10–15 min and washed again with PBS-T. At this point, cells may be used directly for hybridization, or stored for a few days in 1x PBS at 4°C.
For MaSat-, MiSat- and telomere-specific probe hybridization, cells were incubated for 15 min in 0.1 M HCl, washed 3x with 1x PBS. Cells were then and equilibrated for 2 min in 2x SSC and finally for 20 min in 50% formamide/2x SSC. For Alu and LINE-1 FISH, cells were equilibrated in hybridization solution without formamide (see Section 2.5.1). All incubation steps were performed at room temperature.
For metaphase spreads, asynchronously growing cells were treated with colcemid (0.1 µg/ml final concentration) for 1 h, harvested by trypsinization and centrifuged at 500 xg for 5 min. After carefully removing the supernatant, the cell pellet was resuspended in pre-warmed KCl solution (0.075 M at 37°C) and incubated at 37°C for 5 min. To prevent clumping of the cells, the first
1–2 ml were added drop-wise and mixed well by flicking the tube. The rest of the solution was added to give a total volume of approximately 8 ml. After a second centrifugation step, the pellet was fixed by the initial drop-wise addition of freshly prepared fixative solution (methanol:acetic acid, 3:1), which was then filled up to 10 ml. After incubation at 4°C for at least 20 min (overnight incubation is preferred) the samples were again centrifuged and most of the supernatant was removed by decanting. The cell pellet was then finally resuspended in the remaining fixative solution by flicking. The chromosome preparations were made from 15–20 µl drops on microscope slides that were dipped into ddH2O.
For the subsequent hybridization, the slides were air-dried overnight, rehydrated for 10 min in ddH2O and digested at 37°C for 10 min in 0.005% pepsin (P6887, Sigma–Aldrich Chemie GmbH) in 0.01 M HCl. The slides were then dehydrated in 70% and 100% ethanol (5 min each) and air-dried overnight. Before hybridization, slides were equilibrated for 30 min in 50% formamide/2x SSC.
2.5.3 Hybridization
For hybridization of interphase cells, a circle/square the size of the glass coverslip carrying the cells was drawn onto a regular microscope glass slide (Menzel–Gläser Superfrost, Thermo Scientific, Cat. No: ABAA000080##32E) using an ImmunoPen (Calbiochem, Cat. No.: 402176-1EA). The hybridization solution containing the respective probe(s) was then applied to the center of this mark and cells were applied face down onto the hybridization solution. The coverslip was additionally sealed with a silicon ring/square (self-made) and a second microscope slide was applied on top. The whole apparatus was then transferred to a hybridization chamber and tightly sealed (Figure 2B).
For hybridization of metaphase spreads, the area of the slide containing the spreads was encircled using an ImmunoPen. The respective probe(s) were then applied, covered with a coverslip to ensure even distribution of the probe and further sealed with a silicon square and a second microscope slide.
The chamber was subsequently incubated at 80°C for 5 min in a water bath to denature both the genomic DNA and probe. This was followed by incubation in a second water bath at 37°C for 24 h (MaSat, MiSat and telomeres) or overnight at 42°C (Alu and LINE-1).
2.5.4 Washing and Blocking
After hybridization, cells were carefully removed from the microscope slide and washed three times for 5 min with 50% formamide/2x SSC pH 7 at 45°C followed by two washes for 5 min in 2x SSC at 45°C. For the Alu- and LINE-1-specific probes, the washing steps were performed with 2x SSC and 0.1x SSC without formamide. To block non-specific antibody binding sites, cells were incubated for 30 min in 1% BSA/4x SSC at room temperature.
NOTE: For PCNA detection, immunodetection must be performed before continuing with the probe detection (Section 2.6.1 and Figure 2).
2.5.5 Probe Detection
Probes were detected by incubation with a rabbit anti-digoxigenin antibody (1:500, Cat. No.: 700772, Thermo Fisher Scientific), a fluorescently-labeled anti-rabbit IgG antibody or fluorescently-tagged streptavidin diluted in 1% BSA/4x SSC, for 20 min at room temperature and washed three times for 5 min with 0.05% Tween/4x SSC.
A post-fixation step with 1%–4% formaldehyde/PBS for 10 min further stabilized the resulting antibody complexes.
DNA counterstaining was performed by incubating cells for 10 min at room temperature with DAPI (10 μg/ml, 4′,6-diamidin-2-phenylindol) and slides were mounted using Mowiol (Merck KGaA, Cat. No.: 81381-250G).
2.6 Replication Staining
2.6.1 Staining of the Replisome Component PCNA.
Immunofluorescent detection of PCNA usually requires additional fixation with methanol. This step ensures accessibility of the anti-PCNA antibody to the epitope, which is located at the interface between the monomeric subunits of the homotrimeric PCNA ring-complex (Figure S1). However, we noticed that this step could be omitted when the hybridization procedure has been performed before.
To achieve good signals, antibody detection of PCNA must be performed after hybridization and washing but before detection of the probe (Figure 2A). After blocking as described in Section 2.5.4, cells were incubated for 1–2 h at room temperature with an antibody specific for PCNA (mouse anti-PCNA, mAb, DABCO, Cat#: M0879, 1/200) diluted in blocking buffer (1x PBS/0.02% Tween20/2% BSA). After washing (2–3x) in PBS-T (0.02% Tween20), detection with secondary antibodies conjugated with fluorophores suitable for subsequent microscopic detection was performed, before continuing with the probe detection (see Section 2.5.5).
2.6.2 EdU Labeling and Visualization of Replicated DNA
EdU labeling and detection must be carried out before cells are equilibrated in SSC and hybridization is performed.
For detection and staining of active DNA synthesis sites, cells were incubated for 15 min with 10 μM EdU (5-ethynyl-2’-deoxyuridine, Thermo Fisher Scientific, Waltham, MA, USA), washed with 1x PBS and fixed with 3.7% formaldehyde/PBS for 10 min. After washing (3x) with PBS-T (0.02% Tween20), cells were permeabilized with 0.5% Triton-X100/PBS for 15 min and pre-denatured with 0.1 M HCl for 15 min, followed by a second permeabilization step with 0.5% Triton-X100/PBS for 15 min.
EdU was detected using ClickIT chemistry (EdU Click-ROTI kit, Carl Roth, Karlsruhe, Germany) according to the manufacturer's instruction. The 6-FAM and the 3-azido-7-hydroxycoumarin azides were used in final dilutions of 1:1,000 and 1:200, respectively. Cells were post-fixed with 1% formaldehyde/PBS for 10 min before proceeding with equilibration in SSC and hybridization buffer, followed by FISH and probe detection (see sections 2.5.2–2.5.5).
2.7 Confocal Microscopy and Line-Profile Colocalization Analysis
Confocal z-stacks of Repli-FISH stained cells were acquired using the UltraVIEW VoX spinning disc microscopy system (PerkinElmer Life Sciences, Waltham, MA, USA) equipped with an oil immersion 60x Plan Apochromat NA 1.45 objective lens.
To measure and compare the overlap between the major satellite-specific FISH probe signals and the replication signals (EdU), line intensity profile analysis was performed in the mid-focal planes of individual nuclei from cells in different stages of the cell cycle (non-S versus early, mid and late S-phase) using ImageJ (https://imagej.nih.gov/ij/). The intensities of both signals were measured along a line drawn manually through the nuclei and covering both euchromatic and heterochromatic (chromocenters) portions within the cell nucleus. The intensity values of both channels were subsequently plotted against the distance along the line.
2.8 High-Content/High-Throughput Imaging and Analysis
High-content/high-throughput imaging and analysis were performed with the Operetta High-Content Imaging System (Perkin Elmer, Part Number: HH12000000). Multiple fields-of-view (169 in total) were acquired with an immersion free 60x high NA objective (NA 0.9) and automatic image analysis was performed with the associated Harmony software (v3.5). The analysis pipeline involved the initial automatic detection and segmentation of individual cell nuclei based on DAPI counterstaining. Cells overlapping the edge of the image or otherwise deformed cell nuclei were excluded from the analysis. Subsequently, segmentation of major satellite repeats within the individual cell nuclei was performed based on the fluorescence signal of the hybridized MaSat-specific probe. Based on this object segmentation, the fluorescence intensity of the replication signal (PCNA in this case) was measured within all spots per individual cell nucleus. This allowed the comparison of overlapping signals at the different S-phase substages with each other and also with non-S-phase cells (G1- and G2-phase) from the whole population. For this, the cells were sorted according to the different cell cycle stages based on the PCNA patterns in ImageJ using a custom written macro (available upon request). In addition, the DAPI intensity of each nucleus was measured to compare DNA content within the different subpopulations. Boxplots were used to plot (1) the increase in DNA content over the cell cycle (non-S-phase, early and late S-phase) and (2) the accumulation of PCNA at chromocenters in cells at the different cell cycle stages using RStudio (Version 0.99.893).
Harmony analysis pipeline:
-
1) Input image:
- Stack processing: Max. projection
- Flatfield correction: Basic
-
2) Find Nuclei (2):
- Channel: DAPI
- Method A
=> Output population: Nuclei
-
3) Find spots:
- Channel: Alexa 488
- Population: Nuclei
- Region: Nucleus
- Method: A
=> Output population: Alexa488 spots
-
4) Calculate morphological properties:
- Population: Nuclei
- Region: Nucleus
- Method: Standard
-
5) Select population (2):
- Population: Nuclei
- Method: Common filters
- Remove border objects
=> Output population: Nuclei inside [field-of-view]
-
6) Select population:
- Population: Nuclei inside
- Method: Filter by property
- Properties roundness: 0.8
- Properties area [µm2] <300
- Properties area [µm2] >60
=> Output population: Round nuclei inside (exclude deformed nuclei)
-
7) Calculate intensity properties (in whole nucleus and in spots):
- DAPI, MaSat, PCNA
3. Results
Repetitive DNA elements are difficult to investigate using genomic approaches due to their repetitive nature, which hinders their mapping to reference genomes. Here, we describe the use of probes for five different genetic repeat elements in different species (mouse and human) and at different developmental stages for the study of their DNA replication kinetics. The probes presented here are commonly used in our laboratory and were generated via standard procedures (PCR and nick-labeling) as described in Section 2 (Material and Methods). However, we also emphasize the use of other probes that are suitable for the detection of specific DNA sequences and that have been proven useful in cytogenetic studies, e.g. peptide nucleic acids (PNAs; [36]), probes generated via primed in situ synthesis (PRINS; [37,38]) or from bacterial artificial chromosomes (BACs).
In order to confirm their specificity, we performed metaphase FISH with all DNA probes. Figure 4A shows the results for human Alu- and LINE-1-specific probes and their distribution along DAPI-counterstained chromosomes as measured by line-profile colocalization analysis (Figure 4A). Alu elements are abundant in GC-rich regions, whereas LINE-1 elements are predominantly found in AT-rich (GC-poor) regions. We found the distribution of Alu elements correlated negatively with the DAPI staining, which preferantially binds AT-rich regions of the DNA, wheras LINE-1 and DAPI signals correlated positively. Furthermore, double hybridization of mitotic chromosomes with biotin-labeled Alu- and digoxigenin-labeled LINE-1-specific probes showed mutually exclusive binding of the two elements (Figure 4A & Figure 4B), emphasizing their existence in distinct regions of the genome (Figure 1B).
The localization and specificity of the mouse probes against MaSat, MiSat and telomeres were evaluated by triple-color DNA FISH on chromosome preparations. For this, the probes were labeled via PCR using dUTPs conjugated to either digoxigenin-11, biotin-16- or Cy5. The results are summarized in Figure 4C. The telomere probe was found to hybridize specifically to the ends of chromatids, resulting in four signals per chromosome. Major satelite probes displayed only one large signal on each chromosome overlapping with DAPI-intense pericentromeric segments. In contrast, the minor satellite probe hybridization resulted in two distinct spots per chromosome; one per chromatid. In accordance with their chromosomal distribution, the centromeric (MiSat) and pericentromeric (MaSat) DNA signals were found in close proximity. Due to the acrocentric organization of mouse chromosomes (Figure 1A), a similar distribution was observed for proximal telomeres and minor satellite DNA. The clustering of pericentromeric and centromeric heterochromatin as well as the organization of chromosomal ends was also visualized by the simultaneous hybridization of all probes to interphase cells as shown in Figure S2. In summary, these data clearly confirmed the specificity of each probe for its intended target structure.
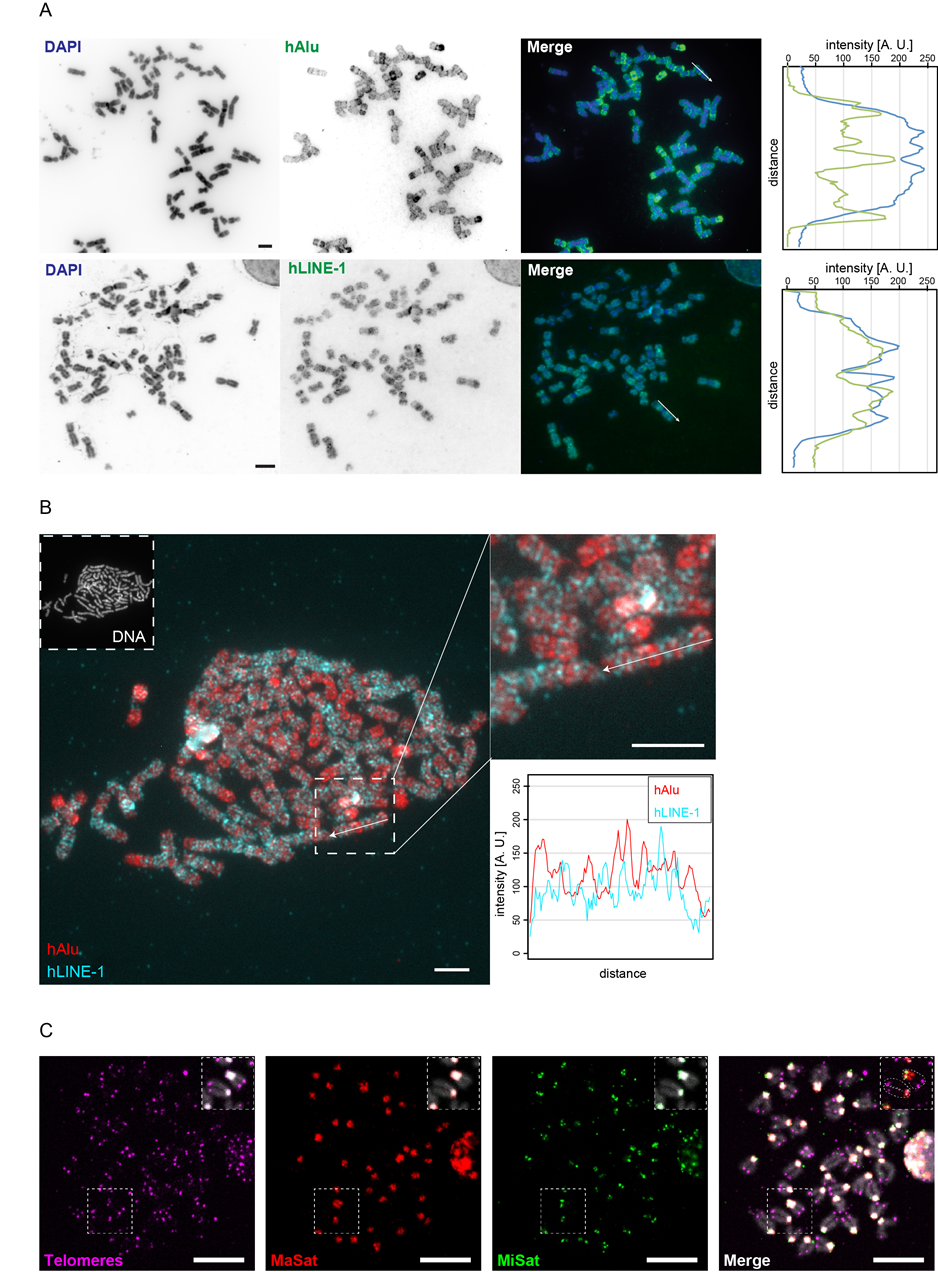
Figure 4 Validation of FISH probe specificity by hybridization to metaphase chromosome preparations. A: Whole metaphase spreads from HeLa cells were hybridized with human Alu-specific (upper row) and human LINE-1-specific (lower row) FISH probes. Chromosomes were counterstained with DAPI. Color line profiles represent the expected overlap between Alu with GC-rich (gene-rich) and LINE-1 with AT-rich (gene-poor) regions, which are differentially stained by DAPI. Scale bar: 5 µm. B: A metaphase spread hybridized to both human Alu-specific (red) and human LINE-1-specific (cyan) probes simultaneously reflects their widespread distribution along eu- and heterochromatic DNA segments (left). The two probes hybridize to mutually exclusive segments along chromosomes as represented by line-profile analysis of a single representative chromosome (right). Scale bar: 5 µm. C: Triple-color FISH combining major satellite-, minor satellite- and telomere-specific probes, each labeled with a different nucleotide analog and hybridized to metaphase chromosome preparations from diploid mouse cells. All probes bind specifically to the chromosomal regions in which the target sequences are organized (see also enlarged insets in each image), i.e. chromosome ends (telomeres, magenta), DAPI-dense pericentromeric regions (MaSat, red) and centromeric regions (MiSat, green). Scale bar: 10 µm.
In both mouse ESC and myoblast interphase nuclei, major satellite DNA was detected as large spots co-localizing with DAPI-intense regions (Figure 5A, upper row). In contrast, minor satellite signals were found in close proximity to the major satellite clusters, forming several distinct spots at their periphery, as expected from their physical location on individual chromosomes (Figure 5A, middle row and Figure 1A). Telomere-specific FISH signals were detected throughout the nucleus, with some localizing adjacent to pericentric heterochromatin (Figure 5A, lower row) due to the acrocentric nature of mouse chromosomes (see Figure 1A).
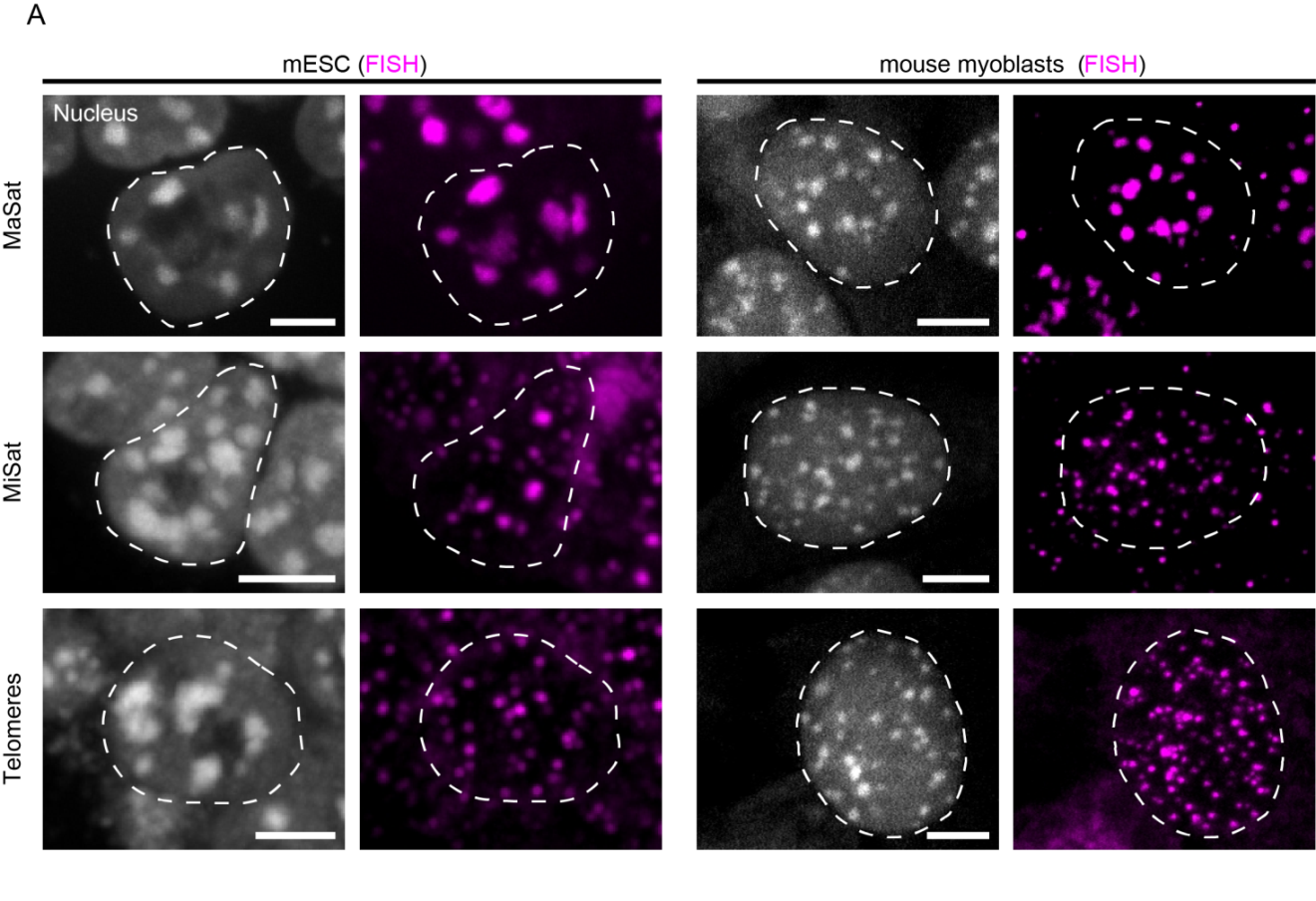
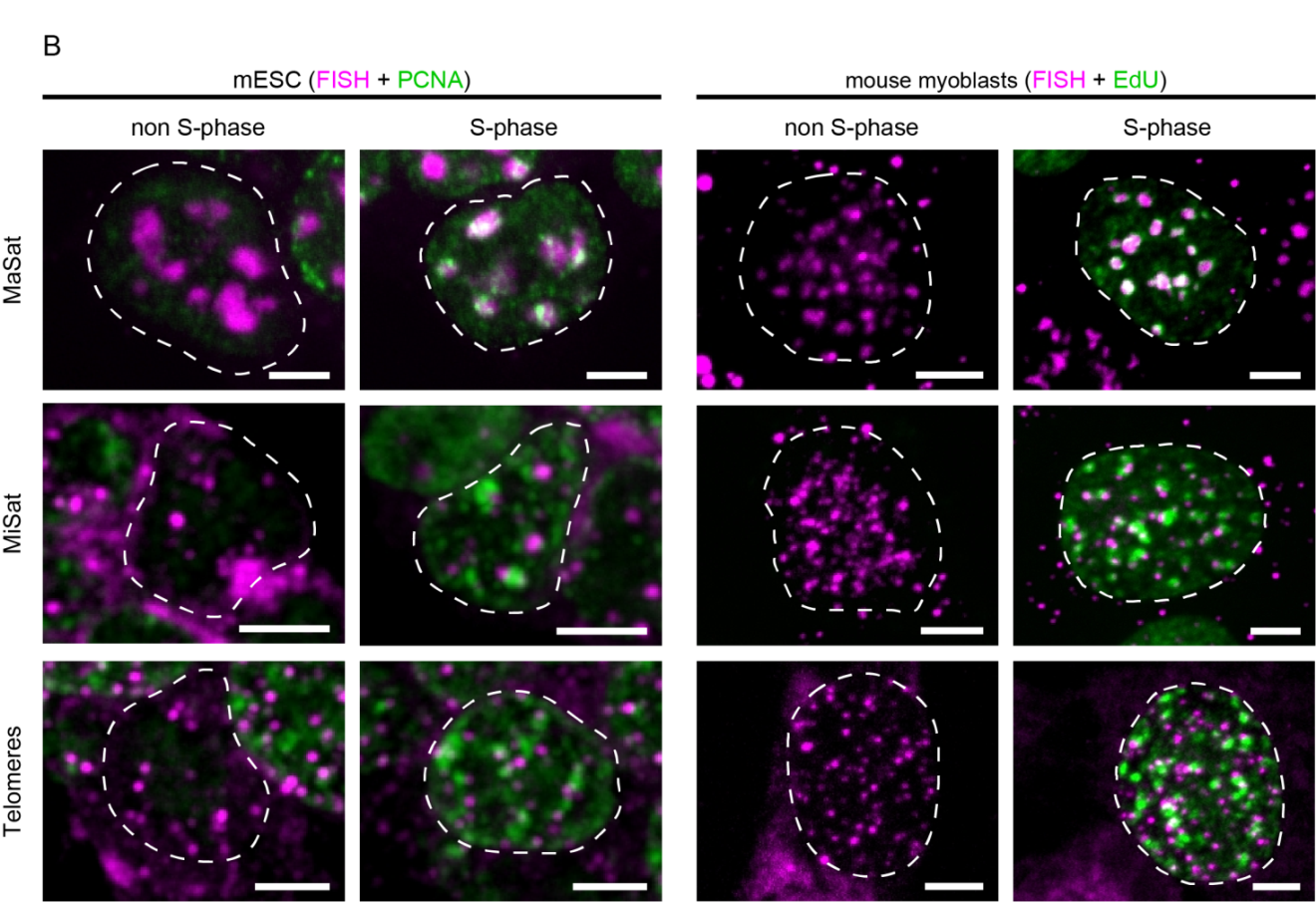
Figure 5 Overview of the localization of MaSat, MiSat and telomeres in murine interphase cells. A: MaSat-, MiSat- and telomere-specific FISH was performed on interphase cells of mouse embryonic stem cells (mESC, left) and mouse myoblasts (right). DNA was visualized by DAPI counterstaining. B: DNA replication was visualized by PCNA co-staining or EdU incorporation and detection. Scale bar: 5 μm.
As interphase chromosomes distribute throughout the nuclear volume, signals for both LINE-1 and Alu interspersed repeats were found throughout the nuclei of human cells, albeit excluded from the nucleoli (Figure 6A). This reflects the fact that both classes of elements cover a large proportion of the genome (e.g. approximately 11% for Alu repeats [39]) as represented by the distribution of LINE-1 and Alu elements on metaphase chromosomes (Figure 1B, Figure 1C and Figure 4B). Notably, the correlation of LINE-1 signals with heterochromatin was stronger than the correlation of Alu signals as judged by the distribution of the DNA and replication signals (note that chromocenters do not form in human cells) (Figure 6A and Figure 6B). A detailed analysis of the distribution of Alu and LINE-1 signals throughout S-phase as well as their replication timing are reported in [40]. In brief, it was shown that Alu elements are replicated during early S-phase, whereas LINE-1 elements are replicated throughout S-phase.
Next, we evaluated the DNA replication kinetics of Alu and LINE-1 elements. We were able to show that both signals, either PCNA or EdU, facilitated discrimination between replicating and non-S-phase cells within an asynchronous cell population (Figure 5B, Figure 6A & Figure 6B). While replicating cells are characterized by the dynamic focal accumulation of the respective signal throughout S-phase, cells in either the G1- or G2-phase of the cell cycle can be identified by the lack of such a pattern. Due to the different types of replication markers used in our approach, non-S-phase cells co-stained for PCNA showed a diffuse nuclear signal representing the unbound nuclear pool of the protein (Figure 5B, left panel, [41,42,43,44]). In contrast, cells in which nascent DNA is marked by the incorporation of the thymidine analog EdU, were only labeled during S-phase and no signals were detected in the G1 and G2 phases (Figure 5B, right panel). Notably, the nuclear localization of the different repetitive elements did not change within the nucleus of S-phase cells, to which the analysis was restricted as can be seen by comparison of the FISH signal distribution in cells at the different stages of S-phase (Figure 5B). Taken together, our results showed that all probes used in this study were highly specific for their intended target sequence (Figure 4), resulting in clear signals with high signal-to-noise ratio when hybridized to interphase nuclei. Furthermore, both nascent DNA as well as replication factors were co-visualized successfully (Figure 5 & Figure 6).
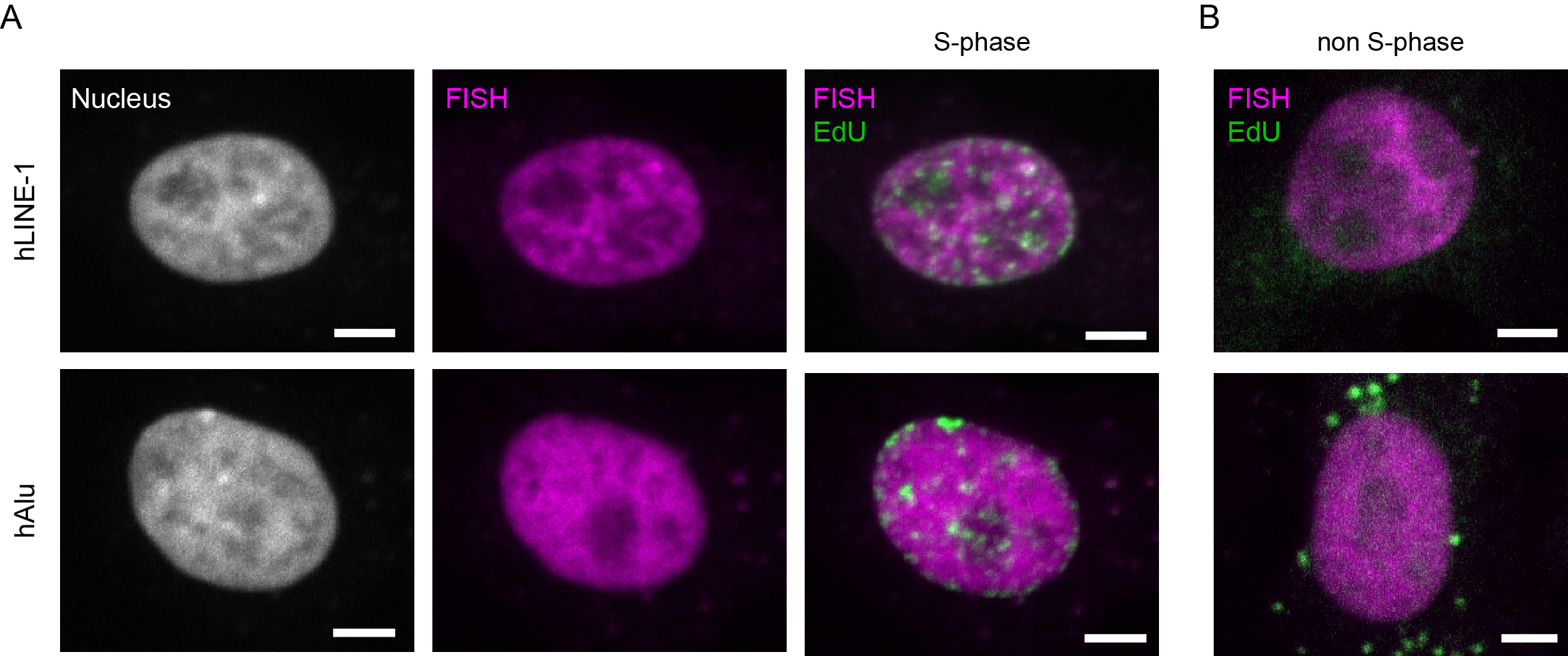
Figure 6 Overview showing the localization of Alu and LINE-1 elements in human interphase cells. A: HeLa interphase cells showing the nuclear distribution of Alu- and LINE-1-specific FISH signals. DNA was visualized by DAPI counterstaining. Alu- and LINE-1-specific FISH signals visualized in combination with EdU in S-phase cells. B: The same probes were observed in cells that are not in S-phase for comparison of their respective distribution in the cell nucleus. Scale bar: 5 μm.
Colocalization analysis (e.g., color line-profile analysis) of the overlapping FISH and replication signals serve as a measure of ongoing DNA replication during the substages of S-phase and, thus, indicate replication timing. Figure 7 shows the outcome of such representative measurements performed on mouse myoblast cells hybridized with the MaSat-specific probe. Again, non-S-phase and replicating cells can be discriminated by the absence or presence of the EdU signal, respectively. Furthermore, the replication of euchromatin (early S), as well as facultative (mid-S) and constitutive heterochromatin (late S), are represented by the changing EdU patterns. The overlap between FISH and replication signals along the analysis path (white arrow) were strongest at late S-phase, which is in complete accordance with previous reports [45,46]. Figure S3 shows the results from a similar analysis performed with cells hybridized to either MiSat- or telomere-specific probes. Here, we found minor satellite DNA replicated asynchronously throughout S-phase, indicated by the overlap of the MiSat FISH signal and replication foci (EdU) shown by line-profile analysis and similar to the results published by [47]. Furthermore, telomeres were also found to replicate throughout the course of S-phase, which is in accordance with previous findings [48,49,50].
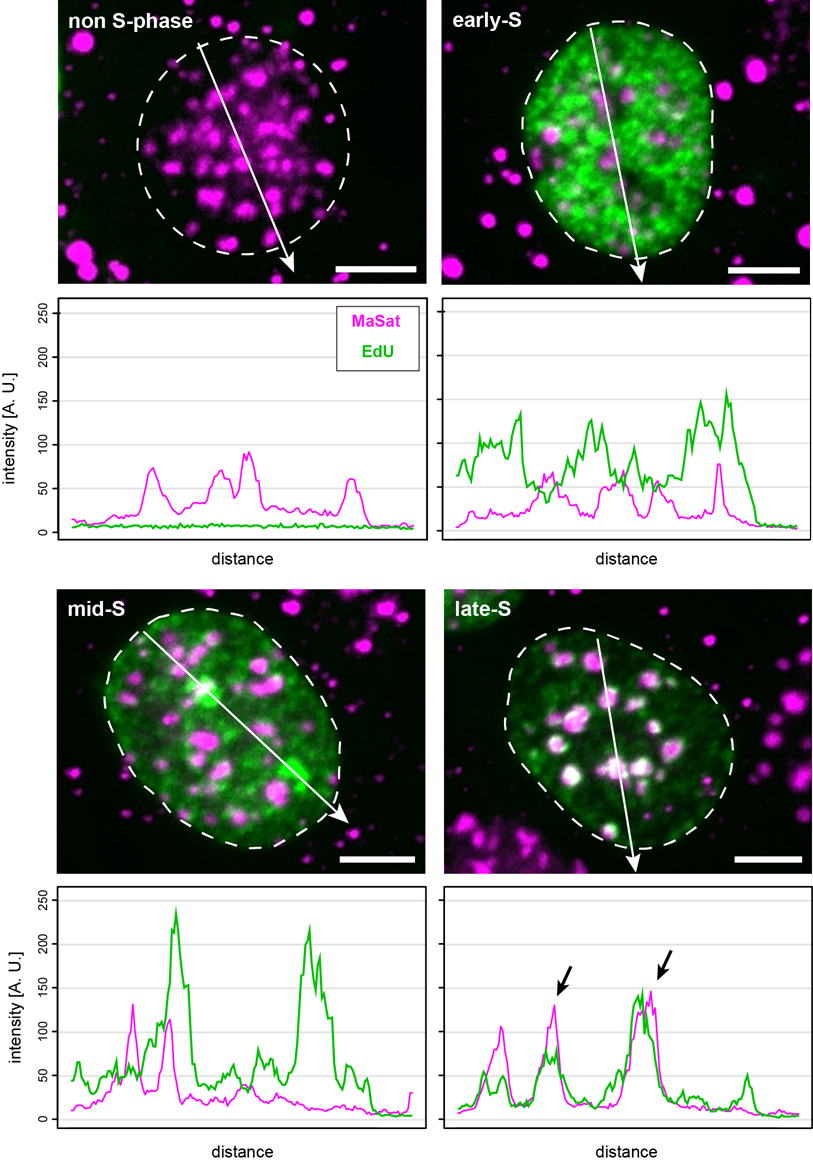
Figure 7 MaSat probe localization in mouse myoblast cells at different S-phase substages. Cells were classified as non-S-phase, early, mid and late replication substage according to their EdU pattern. Colocalization of MaSat FISH (magenta) and replication (green) was analyzed via a color line-profile (black arrows). Scale bar: 5 μm.
These results were also validated and extended by high-content/high-throughput imaging and analysis of asynchronous cell populations (Figure 8). Here, suitable cells were automatically identified based on DNA counterstaining with DAPI (Figure 8A, left). The progression through S-phase was reflected not only by the replication signal pattern, but also by the increase in genomic DNA content over time as measured by the increasing DAPI intensity from cells at the different S-phase substages (Figure 8A, right). The broader signal distribution of non-S-phase cells can be explained by the fact that cells from both G1- and G2-phases of the cell cycle were grouped in this fraction. Cells were classified as non-S-phase cells and cells at either early or late S-phase based on the replication signal distribution (Figure 8B, left). The latter showed a similar increase in the replication signal overlap (PCNA) with major satellite repeats at late S-phase (Figure 8B, right), thus, supporting the previous results of individual cell measurements (Figure 7).
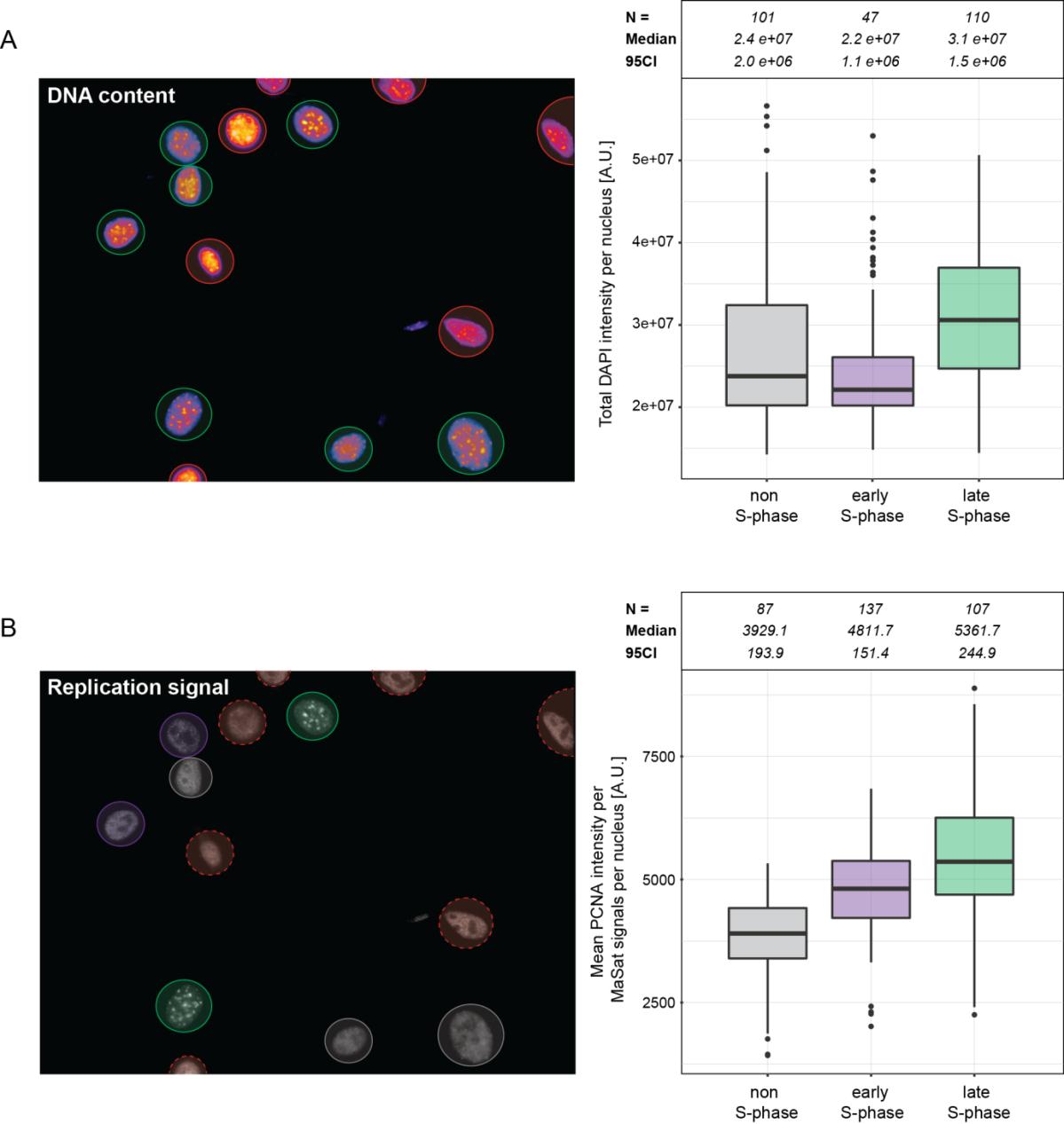
Figure 8 High-content/high-throughput Repli-FISH analysis of asynchronous cell populations. MEF W8 cells hybridized with a major satellite-specific probe and co-stained for PCNA were imaged with a high-content imaging system (Operetta High-content Imaging System, Perkin Elmer, 60x 0.9 NA air objective). A: Image analysis was performed with the Harmony software. Automatic segmentation of cell nuclei was performed based on DNA counterstaining with DAPI (left, green circles). Cells out of focus or on the edge of the image were excluded from the analysis (left, red circles). Increasing DNA content during S-phase is represented by the increase in total DAPI intensity measured in cells at early or late S-phase compared to non-S-phase cells (right). B: Cells were classified by the PCNA signal distribution and sorted into non-S-phase cells (left, gray circles) and cells at either early (left, purple circles) or late S-phase (left, green circles). Comparison of the mean PCNA intensity within all major satellite spots per cell nucleus showed a clear increase toward late S-phase (right, green boxplot), allowing discrimination from the other classes in the population and confirmed the results of individual cell measurements shown in Figure 7.
4. Discussion
Modern next-generation sequencing methods have become increasingly cost and time-efficient. However, available software is still challenged by the correct mapping of repetitive elements both in resequencing and de novo assembly projects. This can be partially explained by the read-length that is now achievable (50–150 bp by next-generation sequencing compared to 800–900 bp obtained by Sanger sequencing), which is substantially shorter than the length of most repeats covering the genome. This results in reads that map to multiple locations within the genome (multi-reads) in resequencing experiments or gaps in de novo genome assemblies [39].
At the same time, several methods have been developed that combine the isolation of nascent DNA from replicating cells with subsequent deep-sequencing (Repli-seq) of the underlying DNA sequence [51]. This method can be used to study the replication timing from cell populations on a genome-wide scale and further offers the ability to relate the sequence information to underlying DNA and chromatin modifications, providing correlative information on the mechanism by which DNA replication is regulated.
Due to the length of time required to sufficiently label and precipitate hundreds of kilobases of labeled DNA (>60 min), these methods have a rather poor temporal resolution. Because of cell population averaging, the analysis focuses mainly on early versus late replicating cells, thereby masking intermediate replication states. Thus, given the fast underlying kinetics and potential variability of the replication process in individual cells, not all questions can be addressed with such methods. In addition, this method is problematic in the alignment of repetitive elements to a reference sequence as described previously.
Although described specifically for different repetitive elements in mouse and human cell lines, the Repli-FISH method can be easily extended using other probes, such as satellite repeat elements [40], rDNA repeat loci [52] or individual genes, e.g. via labeled bacterial artificial chromosomes (BACs) with sequence complementarity to specific genes [53]. In addition, the hybridization procedure can be combined with the detection of other signals of interest, including specific proteins and histone or DNA modifications.
Finally, individual cells can be analyzed either in 3-dimensions at the single cell level or using high-throughput/high-content microscopy techniques and at all levels of microscopical resolution (wide-field, confocal and super-resolution). Of note, the procedure described in this report preserves nuclear integrity (Figure S4), similar to other FISH procedures that include, for example, repeated freezing and thawing in liquid nitrogen [28].
In summary, the Repli-FISH method described here is an efficient and complementary approach to the study of replication kinetics of a broad range of DNA sequences that are otherwise difficult to map and study with the currently available next-generation sequencing methods.
Acknowledgments
We are indebted to Anne Lehmkuhl and Stephanie Meyer for excellent technical assistance. We thank John V. Morgan (U. Michigan Medical School, USA) for providing the pLRE3-eGFP plasmid.
Additional Materials
The following additional materials are upload at the page of this paper.
-
Figure S1: The epitope for the monoclonal anti-PCNA antibody (PC10) maps to the interface of the PCNA monomers.
-
Figure S2: Triple-color FISH combined with replication staining.
-
Figure S3: Localization of MiSat- and telomere-specific probes in mouse myoblast cells at different S-phase substages.
-
Figure S4: Effect of the hybridization procedure on nuclear integrity.
Author Contributions
C.R., P.W. and M.C.C. wrote the manuscript. A.S. performed experiments in HeLa cells with Alu and LINE-1 specific probes. C.R. and P.W. performed experiments in mouse myoblast and ES cells, respectively using probes for major, minor and telomeric repeats and in metaphase cells. C.R. and P.W. analyzed the data.
Funding
This work was supported by grants of the Deutsche Forschungsgemeinschaft (GRK1657/TP1B and DFG CA198/9-1 and 2) to M.C.C..
Competing Interests
The authors have declared that no competing interests exist.
References
- Chagin VO, Stear JH, Cardoso MC. Organization of DNA replication. Csh Perspect Biol. 2010; 2: a000737. [CrossRef]
- Hiratani I, Ryba T, Itoh M, Yokochi T, Schwaiger M, Chang CW, et al. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 2008; 6: e245. [CrossRef]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. 2001; 409: 860-921. [CrossRef]
- Mouse Genome Sequencing C, Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002; 420: 520-562. [CrossRef]
- Batzer MA, Deininger PL. Alu repeats and human genomic diversity. Nat Rev Genet. 2002; 3: 370-379. [CrossRef]
- Korenberg JR, Rykowski MC. Human genome organization: alu, lines and the molecular structure of metaphase chromosome bands. Cell. 1988; 53: 391-400. [CrossRef]
- Ostertag EM, Kazazian HH, Jr. Biology of mammalian L1 retrotransposons. Annu Rev Genet. 2001; 35: 501-538. [CrossRef]
- Martin SL, Bushman FD. Nucleic acid chaperone activity of the ORF1 protein from the mouse LINE-1 retrotransposon. Mol Cell Biol. 2001; 21: 467-475. [CrossRef]
- Kazazian HH, Jr., Moran JV. The impact of L1 retrotransposons on the human genome. Nat Genet. 1998; 19: 19-24. [CrossRef]
- Chu WM, Liu WM, Schmid CW. RNA polymerase III promoter and terminator elements affect Alu RNA expression. Nucleic Acids Res. 1995; 23: 1750-1757. [CrossRef]
- Deininger P. Alu elements: know the SINEs. Genome Biol. 2011; 12: 236. [CrossRef]
- Dewannieux M, Esnault C, Heidmann T. LINE-mediated retrotransposition of marked Alu sequences. Nat Genet. 2003; 35: 41-48. [CrossRef]
- Moyzis RK, Buckingham JM, Cram LS, Dani M, Deaven LL, Jones MD, et al. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc Natl Acad Sci USA. 1988; 85: 6622-6626. [CrossRef]
- de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005; 19: 2100-2110. [CrossRef]
- Wong AK, Rattner JB. Sequence organization and cytological localization of the minor satellite of mouse. Nucleic Acids Res. 1988; 16: 11645-11661. [CrossRef]
- Joseph A, Mitchell AR, Miller OJ. The organization of the mouse satellite DNA at centromeres. Exp Cell Res. 1989; 183: 494-500. [CrossRef]
- Ohno S. So much “junk” DNA in our genome. Brookhaven Symp Biol. 1972; 23: 366-370.
- Orgel LE, Crick FH. Selfish DNA: the ultimate parasite. Nature. 1980; 284: 604-607. [CrossRef]
- Doolittle WF, Sapienza C. Selfish genes, the phenotype paradigm and genome evolution. Nature. 1980; 284: 601-603. [CrossRef]
- Choo KHA. Domain organization at the centromere and neocentromere. Dev Cell. 2001; 1: 165-177. [CrossRef]
- Lyon MF. Do LINEs have a role in X-chromosome inactivation? J Biomed Biotechnol. 2006. [CrossRef]
- Rubin CM, Kimura RH, Schmid CW. Selective stimulation of translational expression by Alu RNA. Nucleic Acids Res. 2002; 30: 3253-3261. [CrossRef]
- Slotkin RK, Martienssen R. Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet. 2007; 8: 272-285. [CrossRef]
- Gey GO, Coffman WD, Kubicek MT. Tissue culture studies of the proliferative capacity of cervical carcinoma and normal epithelium. Cancer Res. 1952: 264–265.
- Yaffe D, Saxel O. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature. 1977; 270: 725-727. [CrossRef]
- Peters AH, O'Carroll D, Scherthan H, Mechtler K, Sauer S, Schofer C, et al. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell. 2001; 107: 323-337. [CrossRef]
- Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992; 69: 915-926. [CrossRef]
- Cremer M, Grasser F, Lanctot C, Muller S, Neusser M, Zinner R, et al. Multicolor 3D fluorescence in situ hybridization for imaging interphase chromosomes. Methods Mol Biol. 2008; 463: 205-239. [CrossRef]
- Liu P, Siciliano J, Seong D, Craig J, Zhao Y, de Jong PJ, et al. Dual Alu polymerase chain reaction primers and conditions for isolation of human chromosome painting probes from hybrid cells. Cancer Genet Cytogenet. 1993; 65: 93-99. [CrossRef]
- Frauer C, Rottach A, Meilinger D, Bultmann S, Fellinger K, Hasenoder S, et al. Different binding properties and function of CXXC zinc finger domains in Dnmt1 and Tet1. PLoS One. 2011; 6: e16627. [CrossRef]
- Lehnertz B, Ueda Y, Derijck AA, Braunschweig U, Perez-Burgos L, Kubicek S, et al. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr Biol. 2003; 13: 1192-1200. [CrossRef]
- Anton T, Bultmann S, Leonhardt H, Markaki Y. Visualization of specific DNA sequences in living mouse embryonic stem cells with a programmable fluorescent CRISPR/Cas system. Nucleus. 2014; 5: 163-172. [CrossRef]
- Garcia-Perez JL, Morell M, Scheys JO, Kulpa DA, Morell S, Carter CC, et al. Epigenetic silencing of engineered L1 retrotransposition events in human embryonic carcinoma cells. Nature. 2010; 466: 769-773. [CrossRef]
- Celeda D, Aldinger K, Haar FM, Hausmann M, Durm M, Ludwig H, et al. Rapid fluorescence in situ hybridization with repetitive DNA probes: quantification by digital image analysis. Cytometry. 1994; 17: 13-25. [CrossRef]
- Celeda D, Bettag U, Cremer C. A simplified combination of DNA probe preparation and fluorescence in situ hybridization. Z Naturforsch C. 1992; 47: 739-747. [CrossRef]
- Paulasova P, Pellestor F. The peptide nucleic acids (PNAs): a new generation of probes for genetic and cytogenetic analyses. Ann Genet. 2004; 47: 349-358. [CrossRef]
- Koch JE, Kolvraa S, Petersen KB, Gregersen N, Bolund L. Oligonucleotide-priming methods for the chromosome-specific labelling of alpha satellite DNA in situ. Chromosoma. 1989; 98: 259-265. [CrossRef]
- Pellestor F. PRINS and in situ PCR protocols. Totowa, NJ: Humana Press; 2006.
- Treangen TJ, Salzberg SL. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet. 2011; 13: 36-46. [CrossRef]
- Natale F, Scholl A, Rapp A, Yu W, Rausch C, Cardoso MC. DNA replication and repair kinetics of Alu, LINE-1 and satellite III genomic repetitive elements. Epigenetics Chromatin. 2018; 11: 61. [CrossRef]
- Bravo R, Frank R, Blundell PA, Macdonald-Bravo H. Cyclin/PCNA is the auxiliary protein of DNA polymerase-delta. Nature. 1987; 326: 515-517. [CrossRef]
- Celis JE, Madsen P. Increased nuclear cyclin/PCNA antigen staining of non S-phase transformed human amnion cells engaged in nucleotide excision DNA repair. FEBS Lett. 1986; 209: 277-283. [CrossRef]
- Celis JE, Madsen P, Nielsen S, Celis A. Nuclear patterns of cyclin (PCNA) antigen distribution subdivide S-phase in cultured cells--some applications of PCNA antibodies. Leuk Res. 1986; 10: 237-249. [CrossRef]
- Leonhardt H, Rahn HP, Weinzierl P, Sporbert A, Cremer T, Zink D, et al. Dynamics of DNA replication factories in living cells. J Cell Biol. 2000; 149: 271-280. [CrossRef]
- O’Keefe RT, Henderson SC, Spector DL. Dynamic organization of DNA replication in mammalian cell nuclei: spatially and temporally defined replication of chromosome-specific alpha-satellite DNA sequences. J Cell Biol. 1992; 116: 1095-1110. [CrossRef]
- Casas-Delucchi CS, van Bemmel JG, Haase S, Herce HD, Nowak D, Meilinger D, et al. Histone hypoacetylation is required to maintain late replication timing of constitutive heterochromatin. Nucleic Acids Res. 2012; 40: 159-169. [CrossRef]
- Weidtkamp-Peters S, Rahn HP, Cardoso MC, Hemmerich P. Replication of centromeric heterochromatin in mouse fibroblasts takes place in early, middle, and late S phase. Histochem Cell Biol. 2006; 125: 91-102. [CrossRef]
- Arnoult N, Schluth-Bolard C, Letessier A, Drascovic I, Bouarich-Bourimi R, Campisi J, et al. Replication timing of human telomeres is chromosome arm-specific, influenced by subtelomeric structures and connected to nuclear localization. PLoS Genet. 2010; 6: e1000920. [CrossRef]
- Wright WE, Tesmer VM, Liao ML, Shay JW. Normal human telomeres are not late replicating. Exp Cell Res. 1999; 251: 492-499. [CrossRef]
- Zou Y, Gryaznov SM, Shay JW, Wright WE, Cornforth MN. Asynchronous replication timing of telomeres at opposite arms of mammalian chromosomes. Proc Natl Acad Sci USA. 2004; 101: 12928-12933. [CrossRef]
- Marchal C, Sasaki T, Vera D, Wilson K, Sima J, Rivera-Mulia JC, et al. Genome-wide analysis of replication timing by next-generation sequencing with E/L Repli-seq. Nat Protoc. 2018; 13: 819-839. [CrossRef]
- Dimitrova DS. DNA replication initiation patterns and spatial dynamics of the human ribosomal RNA gene loci. J Cell Sci. 2011; 124: 2743-2752. [CrossRef]
- Shimizu N, Shingaki K. Macroscopic folding and replication of the homogeneously staining region in late S phase leads to the appearance of replication bands in mitotic chromosomes. J Cell Sci. 2004; 117: 5303-5312. [CrossRef]
- Waseem NH, Lane DP. Monoclonal antibody analysis of the proliferating cell nuclear antigen (PCNA). Structural conservation and the detection of a nucleolar form. J Cell Sci. 1990; 96: 121-129.
- Roos G, Landberg G, Huff JP, Houghten R, Takasaki Y, Tan EM. Analysis of the epitopes of proliferating cell nuclear antigen recognized by monoclonal antibodies. Lab Invest. 1993; 68: 204-210.
