

Current Understanding of DNA Methylation and Age-related Disease
Eunise M. Aquino ![]() , Miles C. Benton †
, Miles C. Benton †![]() , Larisa M. Haupt
, Larisa M. Haupt ![]() , Heidi G. Sutherland
, Heidi G. Sutherland ![]() , Lyn R. Griffiths
, Lyn R. Griffiths ![]() ,
,
Rodney A. Lea †,*![]()
Genomics Research Centre, Institute of Health and Biomedical Innovation, School of Biomedical Sciences, Queensland University of Technology, Brisbane, Australia
† These authors contributed equally to this work.
* Correspondence: Rodney A. Lea ![]() Tel: +61 477933479
Tel: +61 477933479
Received: February 12, 2018 | Accepted: April 02, 2018 | Published: April 12, 2018
OBM Genetics 2018, volume 2, issue 2 doi:10.21926/obm.genet.1802016
Academic Editors: Stéphane Viville and Marcel Mannens
Special Issue: Epigenetic Mechanisms in Health and Disease
Recommended citation: Aquino EM, Benton MC, Haupt LM, Sutherland HG, Griffiths LR, Lea RA. Current Understanding of DNA Methylation and Age-related Disease. OBM Genetics 2018;2(2):016; doi:10.21926/obm.genet.1802016.
© 2018 by the authors. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
Abstract
DNA methylation involves the covalent transfer of a methyl group to the C-5 position of the cytosine ring on a DNA strand. DNA methylation is both heritable and modifiable and can affect gene expression. In recent years, epigenome-wide association studies using high-throughput technologies have associated variation in DNA methylation levels with normal and pathological aging processes in human populations. DNA methylation patterns have been used to construct epigenetic clocks which can serve as potential biomarkers of age-related diseases. Age acceleration, as determined using these epigenetic clocks, has been strongly linked to common diseases including cancer, neurodegenerative diseases, metabolic diseases, and cardiovascular diseases. Identification of these robust associations between DNA methylation and aging may provide new potential therapeutic avenues for preventing and treating age-related diseases. This review focuses on the role of DNA methylation in aging processes and recent advances in epigenome-wide association studies (EWASs) reporting associations between DNA methylation and age-related diseases.
Keywords
Epigenetics; DNA methylation; aging; EWAS; diseases; epigenetic therapies
1. Epigenetics
The definition of epigenetics has been modified over decades. In the mid-twentieth century, epigenetics was defined as the development of a complex organism from a zygote and all processes within [1,2]. With advancing knowledge of the various mechanisms of cellular development and evolution, the definition of epigenetics has altered. Today, epigenetics is more narrowly defined as structural and chemical changes of DNA and associated regulatory proteins (eg. histones), excluding changes to the nucleotide sequences [1,2].
Epigenetic modifications may alter the accessibility to DNA and chromatin. These may involve the recruitment or exclusion of various factors that regulate gene transcription. There are multiple epigenetic mechanisms known to affect these changes. One mechanism, proposed in two independent papers by Riggs [3] and Holliday and Pugh [4], outlined a molecular model in which the cytosine rings of DNA are methylated and demethylated, providing an on/off switch for gene expression [2,5]. DNA methylation also provided an explanation for the heritability of gene expression through somatic cell divisions [5].
Another epigenetic mechanism contributing to the regulation of gene expression involves histone variants and modifications, which alter the chromatin structure and subsequent access of transcription machinery. Histones can undergo several modifications including acetylation, methylation, and phosphorylation. Such modifications are followed by nucleosome remodelling which loosens the chromatin to allow binding of transcription machinery (euchromatin) or tightening of the chromatin to prevent binding (heterochromatin) [1].
More recently, several RNAs have been associated with epigenetic modifications and gene silencing. For example, long non-coding RNAs (lncRNAs) are known for their gene silencing effects in imprinting and X-chromosome inactivation [6]. LncRNAs recruit polycomb complexes which modify histones, and RNA-binding proteins to prevent histone deacetylation or promoter association [6]. Additionally, subclasses of small non-coding RNAs (sncRNAs) have been associated with gene silencing [7]. Piwi-interacting RNAs (piRNA) are an example of sncRNAs that repress transposon expression through de novo methylation [6]. There are also studies suggesting that small-interfering RNA (siRNA) target TATA-box sequences, blocking the initiation of transcription without modifying histones or DNA [6]. Together, these mechanisms demonstrate the complex co-operation of different epigenetic mechanisms for the regulation of gene expression.
2. DNA Methylation
DNA methylation is a well-studied epigenetic mechanism in plants and animals. Methylation involves the covalent transfer of a methyl group to the C-5 position of the cytosine ring on a DNA strand [8,9]. In somatic mammalian cells, 98% of DNA methylation occurs at symmetric cytosine/guanine dinucleotides (CpGs), whereas only 75% of DNA methylation in embryonic stem cells (ESCs) occurs on CpGs [8]. A significant proportion of DNA methylation has been detected in non-CG contexts, such as CHG or CHH (where H can be A, T, or C) [10,11].
DNA methylation has been shown to play a role in normal development, genomic imprinting, X-chromosome inactivation, chromosome stability, suppression of repetitive element transcription and regulation of gene transcription. Dysfunction of DNA methylation processes is strongly linked to many diseases, especially cancer [5,8].
DNA methylation is regulated by DNA methyltransferases. In humans there are four - DNMT1, DNMT2, DNMT3a, and DNMT3b - which serve a variety of functions [8,12]. DNMT1 is responsible for the maintenance of DNA methylation patterns in daughter strands during DNA replication and prefers hemimethylated DNA [8]. DNMT2 methylates cytosine-38 in the anticodon loop of the tRNA for aspartic acid, but does not methylate DNA [13]. DNMT3a and DNMT3b prefer unmethylated CpG dinucleotides and are responsible for de novo methylation during development. DNMT3a, DNMT3b, and DNMT1 cooperatively function to initiate and maintain DNA methylation. In addition, DNMT3L is a DNA methyltransferase-like protein which, although lacks the conserved catalytic domain that is common to the DNA methyltransferases, serves an important role in DNA methylation. DNMT3L has been shown to establish maternal methylation imprints, stimulate DNMT3a activity [14], and assist the DNMTs in binding the methyl group donor [8]. Each of these DNMTs are essential for normal mammalian development [15].
Although DNA methylation is chemically and genetically stable, it is a reversible modification that can occur actively or passively. Passive demethylation occurs when there is a lack of maintenance machinery, such as DNMT1, which results in the dilution of methylation during replication [16,17]. In contrast, active demethylation has several proposed mechanisms. However, most have been discredited due to lack of supporting evidence [17,18,19,20,21]. Two currently accepted demethylation mechanisms involve TET-mediated oxidation of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), which is further oxidated to 5-formylcytosine (5fC) and 5-carboxlcytosine (5caC) [22]. These oxidated states may be diluted through replication, a process similar to passive demethylation [17]. Alternatively, 5fC and 5caC may be excised by thymine DNA glycosylase (TDG) and subsequently replaced by an unmodified cytosine through base excision repair (BER) [23]. Active DNA demethylation has been implicated in several biological pathways. This includes pre-implantation and primordial germ cell development [24,25], maintenance and differentiation of mouse embryonic stem cells [26], neuronal functions [27,28], DNA repair and genomic instability [17].
3. Regulation of Gene Expression by DNA Methylation
Initially it was thought that the sole function of DNA methylation was to silence gene expression. As more recent studies have emerged, it has become evident that the effects of DNA methylation vary depending upon the position in the genome at which it occurs [5,29]. For example, methylation of CpGs within transcription start sites (TSS) has been strongly linked to the prevention of transcription initiation [30,31]. Additionally, inappropriate methylation of CpG islands in, or near, transcription start sites can lead to the silencing of tumour suppressing genes [32]. In contrast, methylation within the gene body has been positively correlated with gene expression [33] and may even stimulate elongation and splicing [5]. Centromeres and other repeat regions can also be methylated and has been shown to improve chromosomal and genome stability [34]. Furthermore, somatic and cancer cells become unviable with a total loss of methylation [35,36], yet this effect has not been observed in ESCs [37].
Repressed genes are usually characterised by methylated CpGs within the promoter region, including CpG islands (CGI) [5,38,39]. However, methylation of promoter CGIs is not a characteristic of all repressed genes. It is usually genes that are permanently repressed that have methylated promoter CGIs. Imprinted genes, genes on the inactive X-chromosome and genes that would otherwise be inappropriately expressed in somatic cells - such as those exclusively expressed in germ cells – are examples of this circumstance [5,30]. However, it is still unclear why only a minority of CGIs are methylated. A summary of the role of DNA methylation in gene expression is shown in Figure 1 below.
The role of DNA methylation on gene silencing remains a subject of debate [40]. Regardless, it has been shown that in some circumstances gene transcription cannot be initiated when promoter CGIs are methylated once nucleosomes have formed [5,39]. Increasing evidence indicates that silencing precedes methylation therefore suggesting that DNA methylation has a maintenance role. This was demonstrated in a number of experiments including genes in the inactivated X-chromosome, and genes implicated in cancer [41,42]. Further proof of this mechanism began to be unravelled by Ooi et al. (2007) who showed that de novo methylation in cells that express DNMT3L required DNMT3a, DNMT3L, and nucleosomes. Since active transcription start sites do not have nucleosomes, de novo methylation cannot occur, therefore the TSS, and the gene, have to be inactivated prior to methylation [43]. Additionally, nucleosomes in active genes are marked by the histone modifications H3K4me2 or H3K4me3 and the histone variant H2A.Z, which are resistant to methylation [43]. This model predicts that higher levels of gene expression decrease the likelihood of de novo methylation with evidence of monoallelic methylation on the least expressed allele supporting this prediction [43]. However, these observations have only been studied in cells that express DNMT3L and it is not known whether similar processes occur in cells not expressing DNMT3L. The mechanism by which DNA methylation within the gene promoter prevents binding of transcription factors occurs through chromatin remodelling [44]. Methylated CpGs allow methyl-CpG-binding domain proteins to recruit repressor complexes and modify histones, to form either heterochromatin or euchromatin [44].
CpG sites within the gene body are more scarce but can be extensively methylated [5]. A strong positive correlation exists between active transcription and DNA methylation in gene bodies within the active X-chromosome [45]. Recently it has also been suggested that DNMT3B-mediated gene body methylation represses intragenic promoters [46], so that rather than being a causative activating mechanism, methylation restricts the activation of cryptic promoters within transcribed genes. Furthermore, a role in splicing regulation has been suggested for methylation as exons are more heavily methylated than introns with a transition occurring at exon-intron boundaries as well as being increased in the nucleosomes [39].
4. Epigenome-wide Association Study Designs and Technologies
Epigenome-wide association studies (EWAS) are a commonly used approach for detecting epigenetic changes, mainly DNA methylation, that may be occurring on the genome as a consequence of, or contributing to, a phenotype [47]. EWAS can identify DNA methylation signatures in either case vs. control designs or explore associations with continuous phenotypic traits, allowing insight into potential relationships between the methylome and a phenotype of interest [47]. These studies have promising potential for understanding disease mechanisms and the identification of biomarkers.
Whole-genome bisulphite sequencing (WGBS) remains the current gold standard method for mapping DNA methylation status [48]. However, it is expensive to perform, and parallelization is a major limitation [48,49]. In light of this, some capture techniques have been developed, such as reduced representation bisulfite sequencing (RRBS), which allows a subset of the genome to be targeted thus reducing costs. However, array-based technologies have gained popularity for their user-friendly, cost effective, time efficient, and reliable results. Until recently the Human 450K Methylation BeadChip was the most widely used array due to its high-throughput capabilities and wide coverage of most genes as well as upstream and downstream regions. However, the 450K design does not include some regulatory regions that have been shown to have an effect on transcription levels and phenotypic variation [48]. Illumina have recently improved upon this by introducing the Infinium MethylationEPIC BeadChip (ie. ~850K), which covers >90% of the original CpGs on the 450k BeadChip with an additional 350,000 CpGs in enhancer regions. However, an important limitation of array-based approaches remains in that they are not able to assay DNA methylation at single-base resolution or discern allele-specificity.
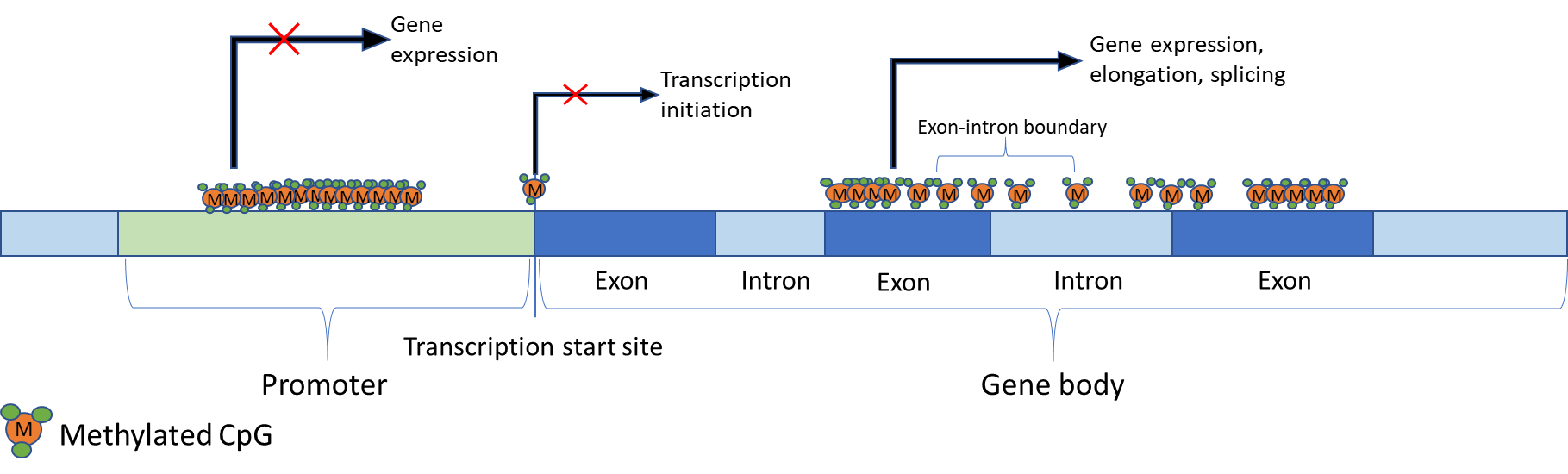
Figure 1 Regulation of gene expression by DNA methylation. Effects of DNA methylation on gene transcription are location-dependent. For example, methylation in the promoter region is observed in repressed genes. Similarly, methylated CpGs on, or near, transcription sites prevent transcription initiation. However, gene body methylation is strongly correlated with gene expression and has been implicated in elongation. Additionally, it has been speculated that gene body methylation may have a role in splicing since exons are more heavily methylated than introns, and there is a transition at exon-intron boundaries.
The choice of EWAS design used is dependent on the aims of the investigation. The simplest approach is the cross-sectional case-control study. This is particularly useful for comparing the epigenome of a group that express a phenotype of interest to those that do not [47,50,51]. Gopalan, et al. [52] used a cross-sectional study design to map and compare DNA methylation patterns across African and European populations to determine whether the patterns are replicated across groups of different genetic and environmental backgrounds. Cross-sectional studies are advantageous due to the large amount of readily available cohorts with DNA samples. However, cross-sectional studies are limited by a number of factors. The epigenome is highly variable, due to genetic factors, but can also be influenced by environmental factors giving rise to inter-individual differences which influence the results. It is also difficult to ascertain whether epigenetic variability is the cause or consequence of the phenotype.
Monozygotic twins are genotypically identical but are not always phenotypically identical. Thus, twin studies are ideal for investigating the effect of the epigenome on phenotype and gene expression [53,54]. Interestingly, one twin study revealed that epigenetic differences naturally arise in individuals as they age, correlating with differential gene expressions in monozygotic twins, demonstrating the robust effect of the epigenome [54]. In addition, twin studies are an ideal EWAS design as it eliminates many confounding factors, particularly environmental and genetic, that are present in non-twin studies. This is especially true for twins raised in similar environments. However, studies investigating causative factors of disease require a longitudinal approach, limiting sample size and resources for appropriate twin studies [55].
Longitudinal and replication studies involve monitoring a study population across a determined time course to track changes in monitored information (phenotypic, genomic, epigenomic, etc), with the goal of inferring associations for phenotypes of interest and potential traits that influence them over time. Such study designs have been used to track changes in DNA methylation associated with healthy aging [38], memory retention [56], diabetic research [57], and cancer research [58], among many other age-related diseases. Ideally, longitudinal studies monitor disease-free, or phenotypic-negative, subjects over many years and observe any phenotypic changes or disease-onset along with DNA methylation changes to identify potential biomarkers [55]. These studies eliminate inter-individual differences that confound results of other EWAS designs and allow observation of changes, rather than identification of differences, in DNA methylation patterns. However, longitudinal studies are expensive, time consuming, and recruiting subjects is often difficult.
As the application of epigenome-wide association studies become broader, more recent studies have been designed using a combination of approaches. This is aimed at reducing, or eliminating, confounding factors to identify more robust associations. Another consideration for EWAS is sample source. Most studies use whole blood, blood components, or saliva, as these are the least invasively obtained and have a wide range of applications including the identification of disease biomarkers in some tissue-specific diseases [59]. Other studies have used other tissue-types, such as brain [60], and cancer tumours [59] where blood is an unsuitable sample. The invasive nature of the procedures required to obtain these samples make these types of studies difficult to conduct. Until less invasive procedures are identified to obtain reliable methylation data, EWAS will be mostly confined to the use of whole-blood, or blood components, and saliva.
As a result, currently, most studies of epigenomics are conducted using mixed cell populations. While this has been very successful, there are limitations in the field that cannot be overcome until epigenomic changes can be observed in single-cell resolution. Recent studies suggest that the use of heterogenous tissues without consideration of cell-subtypes increases false positives [61]. Therefore, deconvolution of signals from mixed cell populations have been recently introduced in epigenetic data analysis. These aim to determine the proportion of cell-subtypes within a sample to reduce false positive associations [62,63]. An alternative to deconvolution of the data sets is the use of single-cell sequencing, a recent advancement in the omics field. It allows the interrogation of all CpG sites in the genome, potentially providing more insightful data into the mechanisms of epigenetic changes and their effect on phenotypic variability in comparison to the current bulk cell approach [64].
5. Epigenetic Clocks - Predictors of Age
The epigenetic drift and epigenetic clock phenomena have provided the basis for epigenetic and aging studies [65,66,67]. Epigenetic drift can be defined as the general decrease in DNA methylation and the increase in variability of methylation patterns with age [67,68]. This has been observed in monozygotic twins, where twin pairs who have similar epigenomes at birth, become more discordant with age [54]. These changes may be in response to environmental influences and some have been significantly linked to phenotypic variability as well as risk of disease. Alternatively, the epigenetic clock describes the changes in DNA methylation status in specific CpG sites across the genomes that are associated with age [67,68,69].
Although there have been several epigenetic predictors of age developed, there are currently only two models that are commonly used. The Hannum [70] and Horvath [59] models were developed independently and use different methods of calculating an individual’s epigenetic age. The Hannum model was developed based on DNA methylation measurements using Illumina 450K array data from whole blood samples of more than 650 individuals from a mixed population [70]. The model identified 71 sites that were highly predictive of chronological age that had a 96% correlation with a margin of error of 3.9 years [70]. In comparison, the predictive model developed by Horvath involved over 7,800 samples from 51 different cell and tissue types. This model identified 353 CpG sites with robust correlations with chronological age. Horvath developed a multi-tissue predictor with a 96% correlation with a margin of error of 3.6 years [59]. Interestingly, epigenetic age estimates determined independently using both models are correlated despite only sharing 6 CpG sites in common [71]. Since epigenetic clocks were derived, they have been used to determine whether epigenetic age is associated with chronological age, and to infer implications on aging.
In addition to developing a predictive model of aging, Horvath also arrived at several other conclusions related to DNA methylation age and aging. He found that certain tissue types produced high error margins when using the predictive model, including breast tissue, skeletal muscle tissue, cardiac tissue, dermal fibroblasts, and uterine endometrium, although reasons for these discrepancies are unclear [59]. The results of this study also demonstrated that the DNA methylation age of both embryonic and induced pluripotent stem cells is near zero. Following from this, Horvath determined that the aging rate of the methylome follows a logarithmic increase until adulthood, then becoming linear thereafter [59].
The study by Horvath also tested the multi-tissue predictor on cancer tissues which exhibited extreme age acceleration [59]. These models have been used in many studies to determine whether lifestyle factors affect age acceleration [51], and whether age acceleration is correlated to disease onset and mortality [50,71].
Other confounders include the changes that occur in blood cell composition with age. Since the majority of epigenetic and aging studies are conducted using whole blood samples, it is important to determine whether the change in blood cell composition affects DNA methylation age. Horvath addressed this issue and concluded that while there is a correlation between DNA methylation ages with the abundance of some blood cell types, the correlation is a confounding effect of chronological age [59]. Additionally, DNA methylation age is not a reflection of changes in blood cell type composition, but rather the intrinsic changes of the methylome [59]. More recent studies have now focussed on the use of two more measures of epigenetic age, namely the intrinsic and extrinsic epigenetic age acceleration (IEAA and EEAA). IEAA measures the epigenetic age in blood independent of blood cell counts affected by chronological age [59,71,72]. In contrast, EEAA uses whole blood samples to measure epigenetic aging of immune cells and changes to blood cell composition [72]. Evidently, the use of IEAA and EEAA are only applicable to blood samples. For all other tissue and cell types, the universal measure of age acceleration is used, which is simply the average epigenetic age compared to chronological age.
6. Age-associated Diseases and DNA Methylation
An aged epigenome is accompanied by changes in DNA methylation which can adversely affect transcriptional processes and downstream protein structure and function [38,54,69,70,73]. Studies examining methylome aging rates and the increase in epigenetic drift revealed that these mechanisms have effects on the risk of age-related disease onset [54,70]. It has been proposed that this increase in disease risk could be due to gradual deregulation of epigenetic mechanisms and regulatory mechanisms. Consequently, this deregulation contributes to the loss of phenotypic plasticity in response to environmental stimuli, promoting aging [74].
A recent EWAS by Zhang, et al. [75] illustrated the role of DNA methylation in mortality. The study investigated DNA methylation in deceased individuals and compared these to a healthy cohort and a validation cohort. From this study, 58 CpG probes were discovered to be strongly associated with mortality and were subsequently replicated in the other cohorts. These CpGs were located across 38 genes, 14 intergenic regions on 19 chromosomes. The genes identified have been linked to a multitude of diseases including diabetes, cardiovascular diseases, cancers, neuropsychiatric disorders, and HIV. Interestingly, none of the CpGs identified were previously associated with age, although the majority were associated with smoking exposure. A risk score was also determined for mortality (independent of cause), cardiovascular diseases, and cancer, based on DNA methylation and identified 10 CpGs to be strong predictors of mortality [75].
Although hypomethylation is usually observed in an aging epigenome, hypermethylation primarily occurs in promoters of genes associated with cancer, such as tumour suppressors [69,74]. This is also observed for CpG islands in genes associated with T-cell immune responses and T-cell differentiation, resulting in decreased expression [76] with hypermethylation linked to the repression of gene expression (model in Figure 2). This likely contributes to the high incidence of cancer and the decline in immunocompetence and response to infection in aging individuals. It is also notable that epigenetic drift in stem cells hinders the repair and regeneration of tissues [77,78], particularly in the gastrointestinal tract and the regeneration of leukocytes. In human somatic cells, the deterioration of methylomes have been observed in central nervous system tissues, specifically in genes related to nervous system development, neuron differentiation, and neurogenesis, eventually leading to neurodegenerative disorders [79]. Hypomethylation of Alu and LINE1 elements during aging has also been related to a significant decline in lung function in the elderly [80]. In addition, CD8+T cells have been identified to have an increased methylome variance associated with age that has been linked to impaired immune responses [76].
Lifestyle factors such as diet, exercise, and cigarette smoking are known to heavily influence the epigenome [51,81]. Exercise affects chromatin dynamics and delays aging while smoking can alter DNA methylation patterns, change the chromatin structure and increase the risk of developing age-related diseases. Diet has also been shown to reverse global and specific DNA methylation in loci correlated with metabolic pathways [81]. Remodelling of genome-wide methylation patterns have been observed as a result of dietary restriction, which delays age-associated changes [82]. Moreover, DNA methylation profiles of calorie-restricted mice and monkeys have showed a lower epigenetic age compared to chronological age [83]. Interestingly, a consistent high nutrient intake appeared to induce age-related methylation in the liver. Some compound in fruits and vegetables has also been shown to have an effect on reducing neurodegenerative and cardiovascular disease as well as chronic inflammation, through mediation of epigenetic processes [74,81].
The CpG site in FTO, a gene associated to fat mass and obesity, has been found to have lower methylation levels in type 2 diabetes mellitus patients when compared to healthy individuals [84]. The insulin gene promoter was also identified to be more hypermethylated in type 2 diabetic patients and negatively correlated with insulin gene expression. In addition, hypermethylation was found to be positively correlated with HbA1c levels [85]. More recently, examination of age acceleration related changes to lifestyle factors including diet, exercise, and education. The study used IEAA and EEAA measures and concluded that a healthy diet, adequate exercise, and higher education were correlated with slower age acceleration, although these effects were not identified to be significant [51].
7. Epigenetic Therapies
The link between epigenetic processes, aging, and age-related diseases has introduced new therapeutic avenues of pursuit. Current epigenetic drugs are generally designed to interfere with histone-modifying enzymes, DNA methyltransferases, and the enzymes that interpret chromatin modifications [86]. DNA methyltransferase inhibitors and histone deacetyltransferase inhibitors are currently two classes of epigenetic drugs commercially available, with several other classes undergoing pre-clinical and clinical phase trials [87].
The mechanism of action of methylation inhibitors, the first class of epigenetic drugs developed, involves inhibition of DNMT1 functions. One example is 5-azacytidine, a cytosine analog that integrates into the DNA strand and forms an irreversible bond with DNMT1, preventing methylation of the daughter strand during replication [88]. A similar drug, zebularine, has been shown to reverse methylation-silencing of genes in mice [89]. More recently, DNA methylation inhibitors have recently been shown to mimic a viral defence pathway by upregulating interferon response in ovarian cancer cells [90].
Antisense oligonucleotides are another means of methylation inhibition. These oligonucleotides bind to the 3’ untranslated region (3’ UTR) of DNMT1 to inhibit the translation of the DNMT1 transcript and have been shown to induce the re-expression of a tumour suppressing gene [91]. In addition to targeting replicating DNA strands and the DNMT1 gene, these small-molecule drugs target the active site of the DNMT1 enzyme, preventing its binding to the target gene [92].
Although the focus to date for many epigenetic drugs has been cancer, the uses of these drugs are currently being explored for other diseases, particularly the neurodegenerative disorders, Alzheimer’s Disease and Parkinson’s Disease [93,94]. Epigenetic drugs present several complex issues as their targets have diverse functions, which further complicate the drug development process [95]. However, new potential drug targets continue to be identified as differentially methylated regions between disease-affected groups and healthy controls are analysed as is occurring in Alzheimer’s Disease and atherosclerosis [96]. Furthermore, studies such as these improve our understanding of these mechanisms in healthy human aging and human longevity.
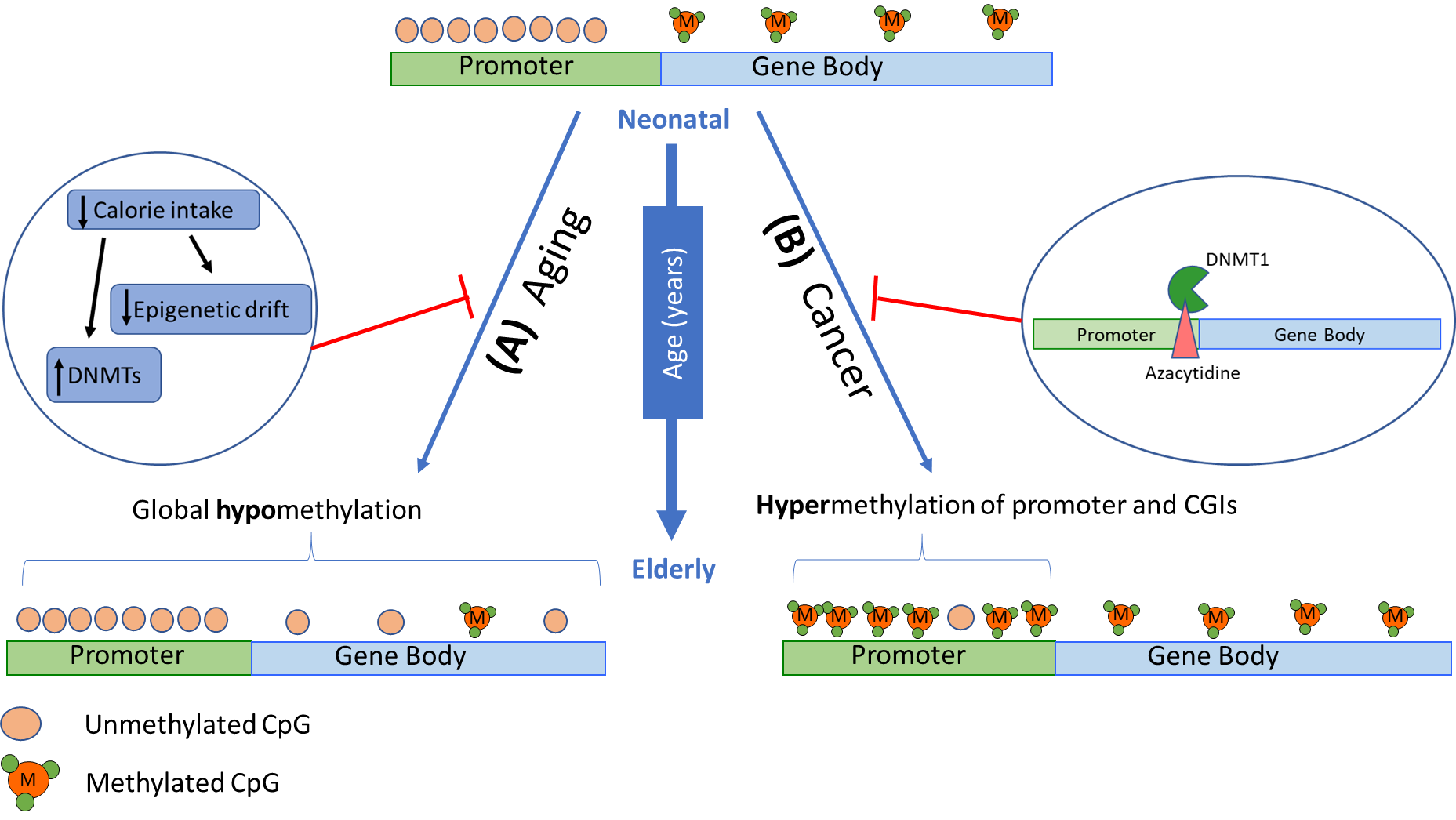
Figure 2 Changes in DNA methylation patterns in a gene with age. DNA methylation patterns have been observed to change with physiological aging and in various age-related diseases. Given an active gene at an early age, CpG sites within gene promoters are typically unmethylated, while gene body CpG sites are methylated. (A) With age, global hypomethylation is usually observed which has been associated with genome instability, hence further promoting aging processes. Lifestyle factors, particularly diet, has been shown to delay age-associated DNA methylation changes and activate DNMTs. (B) Hypermethylation in promoter regions are seen in genes associated with cancer, particularly tumour suppressors, and T-cell immune responses and differentiation. Epigenetic therapies, such as 5-azacytidine, can prevent tumorigenesis by integrating into the DNA sequence of tumour suppressor genes, binding to DNMT1, and preventing inappropriate gene silencing.
8. Conclusions and Future Directions
The mechanisms of DNA methylation are now well documented, including the enzymes responsible for its initiation, maintenance, and regulation. In light of this, focus has now turned to understanding the effects of DNA methylation on gene expression. Studies have shown that methylation of CpG sites in promoter regions are strongly linked to the suppression of transcription, and that methylation of CpG sites within gene bodies are positively correlated to transcription. It has also been observed that methylation of CpG islands is characteristic of some permanently repressed genes, but not all repressed genes have methylated CpG islands. Although these associations are robust and have been demonstrated in numerous studies, the intrinsic mechanisms of these effects are still unclear.
Studies of DNA methylation have included correlating levels at particular genomic loci with phenotypic traits. Recent advancements in array-based technologies have enhanced these studies on a genome wide basis via the use of EWAS. EWAS can utilise a single sample, case-control, longitudinal, twin design or can combine multiple approaches to achieve specific aims. The application of EWAS have been the key to realising the effects of methylation on gene expression, identification of relevant CpG sites to aging, changes to methylation with age, and associations between methylation and age-related diseases. However, current EWAS methods using large cell populations provide only an average of the changes occurring to the methylome. Therefore, progression of epigenomic studies toward single-cell methods to observe specific methylation changes will likely provide significant advances to epigenomic research on stem and cancer cells.
Current epigenetic predictors of age have stemmed from EWAS. Several models have been developed which use different sets of CpGs to determine an epigenetic age. This can then be compared to chronological age to determine any age acceleration. Currently, the Hannum and Horvath models are the most often used for these determinations due to their high correlation and low margins of error. While the Horvath model can be used for multiple cell and tissue types, the Hannum model is more robust when using blood samples. Many studies have concluded strong links between DNA methylation, age acceleration, and increased risk of disease and mortality. While robust, the biological mechanisms behind these associations remain unknown. Although the association between DNA methylation and age-related diseases have introduced new therapeutic avenues, a major challenge exists to overcome target specificity and is the current focus of investigation.
Acknowledgments
The authors thank Mr. Lucas J. Wager for his assistance in refining Figure 2 for this manuscript.
Author Contributions
Eunise Aquino authored the manuscript. Miles Benton, Larisa Haupt, Heidi Sutherland, and Rodney Lea critically revised the manuscript. Lyn Griffiths approved the final version.
Competing Interests
The authors have declared that no competing interests exist.
References
- Felsenfield G. A Brief History of Epigenetics. C SH Perspect Biol, 2014; 4: a108200. [CrossRef]
- Holliday R. Epigenetics: A Historical Overview. Epigenetics. 2006; 1: 76-80. [CrossRef]
- Riggs A. X inactivation, differentiation, and DNA methylation. Cytogenet Cell Genet. 1975; 14: 9-25. [CrossRef]
- Holliday R, Pugh J. DNA Modification mechanisms and gene activity during development. Sci. 1975; 187: 226-232. [CrossRef]
- Jones P. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012; 13: 484-492. [CrossRef]
- Handy D, Castro R, Loscalzo J. Epigenetic modifications: basic mechanisms and role in cardiovascular disease. Circulation. 2011; 123: 2145-2156. [CrossRef]
- Sato F, Tsuchiya S, Meltzer SJ, Shimizu K. MicroRNAs and epigenetics. FEBS J. 2011; 278: 1598-1609. [CrossRef]
- Jin B, Li Y, Robertson K. DNA methylation: superior or subordinate in the epigenetic hierarchy? Gene Canc. 2011; 2: 607-617. [CrossRef]
- Robertson K. DNA methylation, methyltransferases, and cancer. Oncogene. 2001; 20: 3139-3155. [CrossRef]
- Lee J, Jang SJ, Benoit N, Hoque MO, Califano JA, Trink B, et al. Presence of 5-methylcytosine in CpNpG trinucleotides in the human genome. Genomics. 2010; 96: 67-72. [CrossRef]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J. Human DNA methylomes at base resolution show widespread epigenomic differences. Nat. 2009; 462: 315. [CrossRef]
- Lyko F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat Rev Genet. 2017; [Epub ahead of print]. [CrossRef]
- Goll MG, Kirpekar F, Maggert KA, Yoder JA, Hsieh C-L, Zhang X, et al. Methylation of tRNAAsp by the DNA Methyltransferase Homolog Dnmt2. Sci. 2006; 311: 395-398. [CrossRef]
- Chédin F, Lieber MR, Hsieh C-L. The DNA methyltransferase-like protein DNMT3L stimulates de novo methylation by Dnmt3a. P Natl Acad Sci. 2002; 99: 16916. [CrossRef]
- Jin B, Robertson K. DNA Methyltransferases (DNMTs), DNA Damage Repair, and Cancer. Adv Exp Med Biol. 2013; 754: 3-29. [CrossRef]
- He S, Sun H, Lin L, Zhang Y, Chen J, Liang L, et al. Passive DNA demethylation preferentially up-regulates pluripotency-related genes and facilitates the generation of induced pluripotent stem cells. J Biol Chem 2017. [CrossRef]
- Wu X, Zhang Y. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet. 2017; 18: 517. [CrossRef]
- Bhattacharya SK, Ramchandani S, Cervoni N, Szyf M. A mammalian protein with specific demethylase activity for mCpG DNA. Nat. 1999; 397: 579. [CrossRef]
- Aziz Sancar, Laura A. Lindsey-Boltz, Keziban Ünsal-Kaçmaz, Linn S. Molecular Mechanisms of Mammalian DNA Repair and the DNA Damage Checkpoints. Annu Rev Biochem. 2004; 73: 39-85. [CrossRef]
- Morgan HD, Dean W, Coker HA, Petersen-Mahrt SK. Activation-induced Cytidine Deaminase Deaminates 5-Methylcytosine in DNA and Is Expressed in Pluripotent Tissues: Implications for Epigenetic Reprogramming. J Biol Chem. 2004; 279: 52353–52360. [CrossRef]
- Falnes PØ, Johansen RF, Seeberg E. AlkB-mediated oxidative demethylation reverses DNA damage in Escherichia coli. Nat. 2002; 419: 178. [CrossRef]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Sci. 2009; 324: 930. [CrossRef]
- Weber AR, Krawczyk C, Robertson AB, Kuśnierczyk A, Vågbø CB, Schuermann D, et al. Biochemical reconstitution of TET1–TDG–BER-dependent active DNA demethylation reveals a highly coordinated mechanism. Nat Commun. 2016; 7: 10806. [CrossRef]
- Gkountela S, Zhang Kelvin X, Shafiq Tiasha A, Liao W-W, Hargan-Calvopiña J, Chen P-Y, et al. DNA Demethylation Dynamics in the Human Prenatal Germline. Cell. 2015; 161: 1425-1436. [CrossRef]
- Lee Heather J, Hore Timothy A, Reik W. Reprogramming the Methylome: Erasing Memory and Creating Diversity. Cell Stem Cell. 2014; 14: 710-719. [CrossRef]
- Wu SC, Zhang Y. Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol. 2010; 11: 607. [CrossRef]
- Guo JU, Ma DK, Mo H, Ball MP, Jang M-H, Bonaguidi MA, et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat Neurosci. 2011; 14: 1345. [CrossRef]
- Miller CA, Sweatt JD. Covalent Modification of DNA Regulates Memory Formation. Neuron. 2007; 53: 857-869. [CrossRef]
- Schubeler D. Function and information content of DNA methylation. Nat. 2015; 517: 321-326. [CrossRef]
- Medvedeva YA, Khamis AM, Kulakovskiy IV, Ba-Alawi W, Bhuyan MSI, Kawaji H, et al. Effects of cytosine methylation on transcription factor binding sites. BMC Genomics. 2014; 15: 119. [CrossRef]
- Phillips T. The Role of Methylation in Gene Expression. Nat Educ. 2008; 1: 116.
- Stirzaker C, Song JZ, Ng W, Du Q, Armstrong NJ, Locke WJ, et al. Methyl-CpG-binding protein MBD2 plays a key role in maintenance and spread of DNA methylation at CpG islands and shores in cancer. Oncogene. 2017; 36: 1328-1338. [CrossRef]
- Yang X, Han H, De Carvalho Daniel D, Lay Fides D, Jones Peter A, Liang G. Gene Body Methylation Can Alter Gene Expression and Is a Therapeutic Target in Cancer. Cancer Cell. 2014; 26: 577-590. [CrossRef]
- Moarefi A, Chedin F. ICF Syndrome mutations cause a broad spectrum of biochemical defects in DNMT3B-mediated de novo DNA methylation. J Mol Biol. 2011; 409: 758-772. [CrossRef]
- Chen T, Hevi S, Gay F, Tsujimoto N, He T, Zhang B, et al. Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat Genet. 2007; 39: 391. [CrossRef]
- Jackson-Grusby L, Beard C, Possemato R, Tudor M, Fambrough D, Csankovszki G, et al. Loss of genomic methylation causes p53-dependent apoptosis and epigenetic deregulation. Nat Genet. 2001; 27: 31. [CrossRef]
- Tsumura A, Hayakawa T, Kumaki Y, Takebayashi S-i, Sakaue M, Matsuoka C, et al. Maintenance of self-renewal ability of mouse embryonic stem cells in the absence of DNA methyltransferases Dnmt1, Dnmt3a and Dnmt3b. Genes Cells. 2006; 11: 805-814. [CrossRef]
- Benton MC, Sutherland HG, Macartney-Coxson D, Haupt LM, Lea RA, Griffiths LR. Methylome-wide association study of whole blood DNA in the Norfolk Island isolate identifies robust loci associated with age. Aging. 2017; 9: 753-768. [CrossRef]
- Tan Q, Heijmans BT, Hjelmborg JvB, Soerensen M, Christensen K, Christiansen L. Epigenetic drift in the aging genome: a ten-year follow-up in an elderly twin cohort. Int J Epidemiol. 2016; 45: 1146-1158. [CrossRef]
- Bestor TH, Edwards JR, Boulard M. Notes on the role of dynamic DNA methylation in mammalian development. P Natl Acad Sci. 2015; 112: 6796-6799. [CrossRef]
- Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, et al. A Stem Cell-Like Chromatin Pattern May Predispose Tumor Suppressor Genes to DNA Hypermethylation and Silencing in Adult Cancers. Nat genet. 2007; 39: 237-242. [CrossRef]
- Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, et al. Epigenetic stem cell signature in cancer. Nat Genet. 2006; 39: 157. [CrossRef]
- Ooi SKT, Qiu C, Bernstein E, Li K, Jia D, Yang Z, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nat. 2007; 448: 714-717. [CrossRef]
- Lim DH, Maher ER. DNA methylation: a form of epigenetic control of gene expression. Obstet Gynaecol. 2010; 12: 37-42. [CrossRef]
- Joo JE, Novakovic B, Cruickshank M, Doyle LW, Craig JM, Saffrey R. Human active X-specific DNA methylation events showing stability across time and tissues. Eur J Hum Genet. 2014; 22: 1376-1381. [CrossRef]
- Neri F, Rapelli S, Krepelova A, Incarnato D, Parlato C, Basile G, et al. Intragenic DNA methylation prevents spurious transcription initiation. Nat. 2017; 543: 72-77. [CrossRef]
- Birney E, Smith GD, Greally JM. Epigenome-wide Association Studies and the Interpretation of Disease -Omics. PLOS Genet. 2016; 12: e1006105. [CrossRef]
- Pidsley R, Zotenko E, Peters TJ, Lawrence MG, Risbridger GP, Molloy P, et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016; 17: 208. [CrossRef]
- Bibikova M, Lee J, Barnes B, Saedinia-Melnyk S, Zhou L, Shen R, et al. Genome-wide DNA methylation profiling using Infinium assay. Epigenomics. 2009; 1. [CrossRef]
- Horvath S, Pirazzini C, Bacalini MG, Gentilini D, Di Blasio AM, Delledonne M, et al. Decreased epigenetic age of PBMCs from Italian semi-supercentenarians and their offspring. Aging (Albany NY). 2015; 7: 1159-1170. [CrossRef]
- Quach A, Levine ME, Tanaka T, Lu AT, Chen BH, Ferrucci L, et al. Epigenetic clock analysis of diet, exercise, education, and lifestyle factors. Aging (Albany NY). 2017; 9: 419-437. [CrossRef]
- Gopalan S, Carja O, Fagny M, Patin E, Myrick JW, McEwen LM, et al. Trends in DNA Methylation with Age Replicate Across Diverse Human Populations. Genet. 2017; 206: 1659. [CrossRef]
- Christiansen L, Lenart A, Tan Q, Vaupel JW, Aviv A, McGue M, et al. DNA methylation age is associated with mortality in a longitudinal Danish twin study. Aging Cell. 2016; 15: 149-154. [CrossRef]
- Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, et al. Epigenetic differences arise during the lifetime of monozygotic twins. P Natl Acad Sci USA. 2005; 102: 10604-10609. [CrossRef]
- Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-Wide Association Studies for common human diseases. Nat Rev Genet. 2011; 12: 529-541. [CrossRef]
- Degerman S, Josefsson M, Nordin Adolfsson A, Wennstedt S, Landfors M, Haider Z, et al. Maintained memory in aging is associated with young epigenetic age. Neurobiol Aging. 2017; 55: 167-171. [CrossRef]
- Bacos K, Gillberg L, Volkov P, Olsson AH, Hansen T, Pedersen O, et al. Blood-based biomarkers of age-associated epigenetic changes in human islets associate with insulin secretion and diabetes. Nat Commun. 2016; 7: 11089. [CrossRef]
- Vidal E, Sayols S, Moran S, Guillaumet-Adkins A, Schroeder MP, Royo R, et al. A DNA methylation map of human cancer at single base-pair resolution. Oncogene. 2017; 36: 5648-5657. [CrossRef]
- Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013; 14: R115-R115. [CrossRef]
- Horvath S, Mah V, Lu AT, Woo JS, Choi O-W, Jasinska AJ, et al. The cerebellum ages slowly according to the epigenetic clock. Aging (Albany NY). 2015; 7: 294-306. [CrossRef]
- Kennedy D, Lea RA, Benton M. Evaluating linear regression models for deconvolution of methylation signal from mixed blood cell DNA. Manuscript submitted for publication. 2017.
- Houseman EA, Kile ML, Christiani DC, Ince TA, Kelsey KT, Marsit CJ. Reference-free deconvolution of DNA methylation data and mediation by cell composition effects. BMC Bioinformatics. 2016; 17: 259. [CrossRef]
- Jaffe AE, Irizarry RA. Accounting for cell heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014; 15: R31. [CrossRef]
- Schwartzman O, Tanay A. Single-cell epigenomics: techniques and emerging applications. Nat Rev Genet. 2015; 16: 716. [CrossRef]
- Declerck K, Vanden Berghe W. Back to the future: Epigenetic clock plasticity towards healthy aging. Mech Ageing Dev. 2018. [CrossRef]
- Sidler C, Kovalchuk O, Kovalchuk I. Epigenetic Regulation of Cellular Senescence and Aging. Front Genet. 2017; 8: 138. [CrossRef]
- Pérez Raúl F, Tejedor Juan R, Bayón Gustavo F, Fernández Agustín F, Fraga Mario F. Distinct chromatin signatures of DNA hypomethylation in aging and cancer. Aging Cell. 2018; 0: e12744. [CrossRef]
- Jones MJ, Goodman SJ, Kobor MS. DNA methylation and healthy human aging. Aging Cell. 2015; 14: 924-932. [CrossRef]
- Heyn H, Li N, Ferreira HJ, Moran S, Pisano DG, Gomez A, et al. Distinct DNA methylomes of newborns and centenarians. P Natl Acad Sci. 2012; 109: 10522-10527. [CrossRef]
- Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, et al. Genome-wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates. Mol cell. 2013; 49: 359-367. [CrossRef]
- Chen BH, Marioni RE, Colicino E, Peters MJ, Ward-Caviness CK, Tsai P-C, et al. DNA methylation-based measures of biological age: meta-analysis predicting time to death. Aging (Albany NY). 2016; 8: 1844-1859. [CrossRef]
- Horvath S, Gurven M, Levine ME, Trumble BC, Kaplan H, Allayee H, et al. An epigenetic clock analysis of race/ethnicity, sex, and coronary heart disease. Genome Biol. 2016; 17: 171. [CrossRef]
- McClay JL, Aberg KA, Clark SL, Nerella S, Kumar G, Xie LY, et al. A methylome-wide study of aging using massively parallel sequencing of the methyl-CpG-enriched genomic fraction from blood in over 700 subjects. Hum Mol Genet. 2014; 23: 1175-1185. [CrossRef]
- Li Y, Tollefsbol T. Age-related epigenetic drift and phenotypic plasticity loss: implications in prevention of age-related human diseases. Epigenomics. 2016; 8: 1637-1651. [CrossRef]
- Zhang Y, Wilson R, Heiss J, Breitling LP, Saum K-U, Schöttker B, et al. DNA methylation signatures in peripheral blood strongly predict all-cause mortality. Nat Commun. 2017, 8:14617. 2017; 8: 14617.
- Tserel L, Kolde R, Limbach M, Tretyakov K, Kasela S, Kisand K, et al. Age-related profiling of DNA methylation in CD8+ T cells reveals changes in immune response and transcriptional regulator genes. Sci Rep. 2015; 5: 13107. [CrossRef]
- Beerman I, Bock C, Garrison Brian S, Smith Zachary D, Gu H, Meissner A, et al. Proliferation-Dependent Alterations of the DNA Methylation Landscape Underlie Hematopoietic Stem Cell Aging. Cell Stem Cell. 2013; 12: 413-425. [CrossRef]
- Sun D, Luo M, Jeong M, Rodriguez B, Xia Z, Hannah R, et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell stem cell. 2014; 14: 673-688. [CrossRef]
- Xiao F-H, Kong Q-P, Perry B, He Y-H. Progress on the role of DNA methylation in aging and longevity. Brief Funct Genomics. 2016; 15: 454-459. [CrossRef]
- Lange NE, Sordillo J, Tarantini L, Bollati V, Sparrow D, Vokonas P, et al. Alu and LINE-1 methylation and lung function in the normative ageing study. BMJ Open. 2012; 2. [CrossRef]
- Johnson AA, Akman K, Calimport SRG, Wuttke D, Stolzing A, de Magalhães JP. The Role of DNA Methylation in Aging, Rejuvenation, and Age-Related Disease. Rejuv Res. 2012; 15: 483-494. [CrossRef]
- Hahn O, Grönke S, Stubbs TM, Ficz G, Hendrich O, Krueger F, et al. Dietary restriction protects from age-associated DNA methylation and induces epigenetic reprogramming of lipid metabolism. Genome Biol. 2017; 18: 56. [CrossRef]
- Maegawa S, Lu Y, Tahara T, Lee JT, Madzo J, Liang S, et al. Caloric restriction delays age-related methylation drift. Nat Commun. 2017; 8: 539. [CrossRef]
- Toperoff G, Aran D, Kark JD, Rosenberg M, Dubnikov T, Nissan B, et al. Genome-wide survey reveals predisposing diabetes type 2-related DNA methylation variations in human peripheral blood. Hum Mol Genet. 2012; 21: 371-383. [CrossRef]
- Yang BT, Dayeh TA, Kirkpatrick CL, Taneera J, Kumar R, Groop L, et al. Insulin promoter DNA methylation correlates negatively with insulin gene expression and positively with HbA(1c) levels in human pancreatic islets. Diabetologia. 2011; 54: 360-367. [CrossRef]
- Altucci L, Rots MG. Epigenetic drugs: from chemistry via biology to medicine and back. Clin Epigenetics. 2016; 8: 56. [CrossRef]
- Heerboth S, Lapinska K, Snyder N, Leary M, Rollinson S, Sarkar S. Use of Epigenetic Drugs in Disease: An Overview. Genet Epigenet. 2014; 6: 9-19. [CrossRef]
- Füller M, Klein M, Schmidt E, Rohde C, Göllner S, Schulze I, et al. 5-Azacytidine enhances efficacy of multiple chemotherapy drugs in AML and lung cancer with modulation of CpG methylation. Int J Oncol. 2014; 46: 1192-1204. [CrossRef]
- Cheng JC, Matsen CB, Gonzales FA, Ye W, Greer S, Marquez VE, et al. Inhibition of DNA Methylation and Reactivation of Silenced Genes by Zebularine. JNCI: J Natl Cancer I. 2003; 95: 399-409. [CrossRef]
- Chiappinelli Katherine B, Strissel Pamela L, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell. 2015; 162: 974-986. [CrossRef]
- Winquist E, Knox J, Ayoub J-P, Wood L, Wainman N, Reid GK, et al. Phase II trial of DNA methyltransferase 1 inhibition with the antisense oligonucleotide MG98 in patients with metastatic renal carcinoma: A National Cancer Institute of Canada Clinical Trials Group investigational new drug study. Invest New Drug. 2006; 24: 159-167. [CrossRef]
- Savickiene J, Treigyte G, Jonusiene V, Bruzaite R, Borutinskaite V-V, Navakauskiene R. Epigenetic changes by zebularine leading to enhanced differentiation of human promyelocytic leukemia NB4 and KG1 cells. Mol Cell Biochem. 2012; 359. [CrossRef]
- Delgado-Morales R, Agís-Balboa RC, Esteller M, Berdasco M. Epigenetic mechanisms during ageing and neurogenesis as novel therapeutic avenues in human brain disorders. Clin Epigenetics. 2017; 9: 67. [CrossRef]
- Vaiserman AM, Pasyukova EG. Epigenetic drugs: a novel anti-aging strategy? Front Genet. 2012; 3: 224. [CrossRef]
- Licht JD. DNA Methylation Inhibitors in Cancer Therapy: The Immunity Dimension. Cell. 2015; 162: 938-939. [CrossRef]
- Xiao F-H, He Y-H, Li Q-G, Wu H, Luo L-H, Kong Q-P. A Genome-Wide Scan Reveals Important Roles of DNA Methylation in Human Longevity by Regulating Age-Related Disease Genes. PLOS ONE. 2015; 10: e0120388. [CrossRef]

