

Cell Senescence, Telomerase, and Senolytic Therapy
Michael Fossel * ![]()
- Telocyte LLC, 250 Monroe NE, Grand Rapids, MI, USA
* Correspondence: Michael Fossel ![]()
Academic Editor: James S. Powers
Special Issue: Perspectives on Telomeres and Aging
Received: December 31, 2018 | Accepted: January 31, 2019 | Published: February 15, 2019
OBM Geriatrics 2019, Volume 3, Issue 1 doi:10.21926/obm.geriatr.1901034
Recommended citation: Fossel M. Cell Senescence, Telomerase, and Senolytic Therapy. OBM Geriatrics 2019; 3(1): 034; doi:10.21926/obm.geriatr.1901034.
© 2019 by the authors. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
Abstract
The consensus that cell senescence plays a role in age-related disease has prompted a number of potential clinical interventions, including attempts to reset cell senescence and attempts to remove senescent cells from aging tissues. The latter approach, senolytic therapy, has attracted considerable attention, but both theoretical considerations and published data suggest that the clinical benefits will be transient and that senolytic therapies will likely accelerate long-term degenerative disease. We review the overall field, its history, the theoretical aspects, and the available data. The long-term risks are underestimated and based on naïve assumptions, while the long-term benefits are not borne out by physiologic considerations or data. Senolytics are likely to accelerate tissue pathology, exacerbate clinical disease, and result in early morbidity and mortality.
Keywords
Cell senescence; telomeres; telomerase; senolytics
1. Introduction
Recent advances in our understanding of the role of senescent cells in age-related human disease have prompted several distinct interventional strategies, including:
1) Telomerase gene therapy to reset gene expression in senescent cells,
2) Small molecular drugs aimed at individual genes or proteins, and
3) Senolytic drugs to kill senescent cells.
These three approaches will be compared in light of human pathology and available data (generally animal data), with a focus on current data, as well as the clinical implications of senolytic therapy. A conceptual illustration of the point of intervention of each of these three approaches (and currently available symptomatic drugs), is shown here (Figure 1).
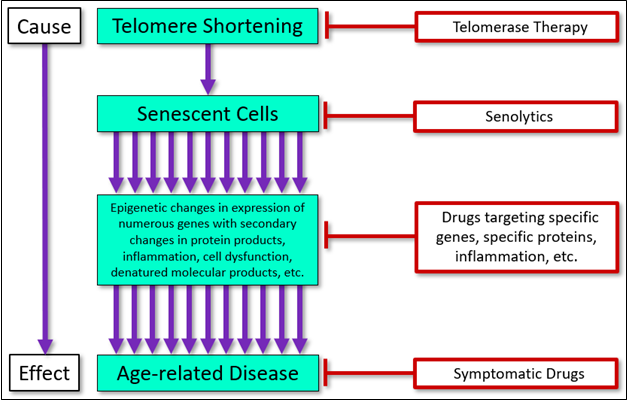
Figure 1 Targets and interventional approaches to age-related disease.
2. Background
Cell senescence was first demonstrated in the 1960’s, predominantly due to the work of Hayflick and Moorhead [1,2]. In 1990, the work of Harley and his colleagues at Geron demonstrated that cell senescence correlated closely with changes in telomere length [3], and this was followed by a series of confirmatory papers [4,5]. Thereafter, the relationship between telomeres and cell senescence was shown to be not only correlational, but causal in a series of papers showing that resetting telomere lengths also reset cell senescence as measured by gene arrays, histology, and cell function [6,7,8].
The first articles in the medical literature [9,10] as well as a textbook on the area [11], suggested that not only was cell senescence a key player in age-related human disease, but that cell senescence in general and telomeres in particular could serve as a uniquely effective point of clinical intervention. In the case of age-related vascular disease, for example, endothelial cell senescence was seen to precede the usual histologic hallmarks of atherosclerosis and do so in a manner that paralleled the locations and degree of pathology [12]. Similar findings were seen in other age-related diseases, such as osteoarthritis and osteoporosis, in which key cells (e.g., chondrocytes and osteocytes) showed senescent changes that preceded and appeared to cause the onset of clinical disease. Such senescent cells included vascular endothelial cells, chondrocytes, osteocytes, and other cell types in their respective tissues and organs.
Over the past two decades, and despite its obvious potential, pursuit of the potential of this initial work was limited by the available technology, which made it difficult to translate this work into in vivo animal or human studies, although some articles continued to clarify the model and point out the clinical possibilities. Specifically, additional experimental support for the model required genetic manipulations that became available as the field matured. In regard to human gene therapy, initial adverse events (e.g., the Jesse Gelsinger fatality in 1999) delayed progress and testing in human gene therapy trials.
Nonetheless, supportive animal and human data contined to accrue. With regard to the putative role of cell senescence in age-related disease, additional findings [13] suggested a similar process was occurring with the aging brain, specifically in glial cells, and that this process might underlie common clinical dementias, such as Alzheimer’s type dementia [14]. Specifically, such glial cells demonstrate telomere shortening and a decreased ability to produce, bind, internalize, and breakdown key molecules, such as beta amyloid [15]. Moreover, glial cell senescence has been seen to precede neurofibrillary tangles and neurodegeneration [16]. Cell senescence is now seen as the cause, rather than the effect, of neural degeneration. As a 2018 editorial in Nature put it: “glial senescence ultimately promotes neuronal degeneration” [17]. The same view, that cell senescence is a causal factor not only in neuronal degeneration specifically, but in age-related diseases generally, is becoming the consensus view.
The first approach mentioned above, resetting gene expression, has worked well in animal studies [18,19], and has solid theoretical support [11], but will not be addressed in detail here, although the use of telomerase to reset gene expression has the clear potential to reverse the disease course in age-related disease. The third approach, the use of small molecules, has shown limited efficacy: while effective in addressing specific genes and proteins, it does not address the broader panoply of changes in genes and proteins that characterize cell senescence and, in consequence, has shown no effect in altering the disease course in age-related disease. The classic example of this failure has been the use of monoclonal antibodies in Alzheimer’s disease trials. The fourth approach, symptomatic therapies, not shown to affect the disease course of age-related disease, is outside of both the interest and the scope of this paper.
The second approach, the use of senolytics will be evaluated here in some detail. We will consider the rationale for its use, the claims that have been made for its clinical potential, the data on its use, and implications of such data for both clinical use and economic value.
3. Rationale for Senolytics
Senescing (as well as fully senescent) cells become increasingly common in older tissues [20]. Such cells demonstrate multiple defining changes, including both generic (characteristic of senescent cells generally) and cell-specific changes (characteristic of specific tissue and cell types). Senescence is associated with telomere-modulated, characteristic changes in gene expression, termed SAGE (senescence associated gene expression). Senescing cells show pronounced intracellular changes including slower molecular turnover, slower DNA repair, deficient cell maintenance, defective mitochondrial function, less efficient ATP production (and a lower ATP/ROS ratio), increased percentages of accrued molecular damage, etc [11]. Nor is this increasing cellular dysfunction limited to intracellular venues. While senescing cells are known to become less effective in performing their own internal cellular functions, they also increasingly interfere in the functioning of other local, non-senescent cells. Bluntly, senescing cells not only cause tissue dysfunction by a passive loss of normal internal function, but they also cause tissue dysfunction by an active and external interference in the function of other cells. This process, termed SASP (senescence associated secretory phenotype) [21,22] is generic to senescing cells in any tissue, with specific characteristics for individual cell and tissue type as well. It is characterized by the secretion of toxic molecules [23,24], as well as by numerous molecular markers and changes in gene expression [25], including p16INK4A, and it can be identified by molecular markers [26], including SA-β-Gal, inflammatory signalling molecules, growth factors and proteases [17].
To take a specific tissue as an example, the chondrocytes making up the bulk of any human joint surface senesce and demonstrate increasing internal cellular dysfunction as they do so. In addition, the dysfunction of such senescent cells actively interferes with the function of other neighboring non-senescent chondrocytic cells as well [23,27]. Osteoarthritis is the clinical result in an aging joint and is characterized by increasing inflammation, as well as the erosion and gross loss of normal joint surface. Current clinical interventions for osteoarthritis include drugs that are marginally or transiently effective (i.e., anti-inflammatory drugs, etc.) for symptomatic treatment or the complete removal and replacement of the affected joint (i.e., surgical replacement with an artificial joint). While replacement has become the orthopedic standard-of- care, it is expensive, painful, debilitating, inherently risky, and does not address the pathology per se. In short, current treatment options are sub-optimal. Generically, the same caveats pertain to all current interventions for age-related disease: current therapies have little-or-no effect upon the disease process itself and are clinically sub-optimal.
As a result, the three basic approaches listed above are all under consideration in the context of age-related disease. Currently available approaches intervene “downstream”, at the level of clinical effects (symptoms) rather than “upstream” at the level of clinical causes (underlying disease processes). Again using the example of the aging joint and its chondrocytes, currently available therapeutic approaches do not address the underlying cause(s) of osteoarthritis, but merely serve to ameliorate symptoms or act by removing the aging tissue in toto. In contrast to current therapy, the three approaches listed above aim to address “upstream” processes at varying levels. Amplifying these three strategies, we may summarize each approach as follows:
1) At the telomere level: telomerase gene therapy aims to reset the telomere length, and hence gene expression, enabling the senescent cells to function normally and abrogating their interference with other, non-senescent cells. Here the goal is to fully restore normal cell and tissue function, restoring the joint to its previous, clinically normal state.
2) At the specific protein and gene level: small molecular drugs aim to individually target any and all characterized gene or molecular changes in the tissue, including cellular debris, mitochondrial changes, free radicals, cellular apoptosis, etc. While the goal is to mitigate the changes (particularly those of SASP), the sheer number of individual changes necessitates dozens of simultaneous interventions, none of which address the underlying cell senescence that drives such changes.
3) At the tissue level: senolytic therapy aims to remove senescent cells, mitigating or preventing SASP effects, and putatively enabling remaining, non- senescent cells to function more normally.
The last approach, removal of senescent cells, is generally termed “senolysis” in the medical and biotechnology literature, and putative agents are termed “senolytics”. Initial data supported a potential value of senolytics [28,29] and several biotechnology companies are currently engaged in promoting this approach [30]. Researchers and biotechnology companies have proclaimed a growing number of putative senolytic compounds, including AP20187 [27], ABT263 [31], INK-ATTAC [29], ABT-263 [32], FOXO4 peptide [33], UBX0101 and UBX1967 [34], and a growing list of others. This approach has garnered considerable interest in the research [35,36] and clinical literature [28,37], as well as among investors and the venture capital community.
4. Claims Regarding Senolytics
A number of articles have suggested that clearance of senescent cells can attenuate age- related tissue changes (or age-related disease) and create a more favorable tissue environment [36,38], and should therefore be considered appropriate for human clinical trials. Such articles accurately stress the likely role of senescent cells in age-related pathology, as well as the factors contributing to senescence, such as age per se, trauma, infection, etc. Published outcomes are generally supportive and intriguing, although (as discussed below) long-term effects are generally not evaluated [23,29,31,33,38] or when long-term data appears in the publication, it is glossed over in favor of the initial positive effects, which are highlighted [27].
Such initially positive effects are consistent, as would be expected from the known effects of senescent cells in aging tissues, and these effects have been documented in several species and in different cell and tissue types. These cell and tissue types encompass several age-related pathologies [36], including tumorigenesis (as well as chemotherapy [33,39]) and age-related deterioration in the kidney, heart, and fatty tissue [27], bone marrow and muscle [31], eye [29], brain [40], etc. These results have prompted further work on specific age-related diseases and prospective human trials (as well as considerable interest in the clinical and investment potential of these approaches).
In osteoarthritis, for example, there is good evidence that senescent cells accumulate in and parallel the course of joint pathology. Moreover, senolysis-attenuated post-traumatic osteoarthritis in mice in vivo and, human in vitro cultures taken from knee replacement patients, showed short-term benefits as assessed by inflammation and matrix protein formation. This suggests a potential benefit in human osteoarthritis [38].
In the case of vascular age-related disease, similar findings are seen (at least in mice), with senescent cells being common in atherosclerotic lesions, including foamy macrophages. Such results suggest that senescent cells play central roles in plaque formation, loss of elasticity, fibrous tissue, plaque rupture, and subsequent clots. The results imply a potential use for senolytic therapy in human atherosclerosis, although interventional data to support the suggestion is lacking [23]. Note that the role of cell senescence in human atherosclerosis was first noted more than two decades ago [12], as was its probable role in atherogenesis [8,41,42,43]. The clinical potential for intervention in age-related arterial disease as well as other age-related disease by addressing cell senescence (using telomerase rather than senolysis) was also pointed out at that time [9,10,44,45,46] and in more detail since [11].
Other proposed clinical targets include ophthalmologic disease as well as age-related diseases of the heart, kidney and liver [47]. Some authors have gone on to argue that the use of senolytics not only has benefits to treating the age-related pathology of specific tissues or organ systems, but will extend the healthy lifespan in animal models and would have similar effects in humans [27,29,31,48]. Although articles advocating senolytic approaches occasionally note the possibility of acceleration of cell division in the remaining, initially non-senescent cells, no attention is given to the adverse consequences (enforced cell senescence of initially non- senescent cells) put into play by senolytic interventions [38].
5. Adverse Effects of Senolytics
Histological considerations suggest that senolytics will accelerate age-related disease in affected tissues. In a sense, this would be parallel to the known effects of radiation, which can induce cell senescence and reduce stem cell availability [49]. In the case of senolytics, the removal of senescent cells would accelerate stem cell division and consequent cell senescence, inducing premature cell and tissue aging, with subsequent acceleration of age-related clinical disease.
In any given tissue, there is a population of cells, some of which are senescent. If we remove cells (e.g., with senolytic therapy), the absence of those cells will trigger the remaining cells to divide in order to replace the cells which are no longer present. This standard, classsical histological response within any tissue replaces lost cells, as occurs with trauma to the dermis, routine epidermal turnover, the death and replacement of circulating lymphocytes, quotidian erythrocyte loss and replacement, hepatocyte turnover, etc. As these cells divide and replace cellular losses, their telomeres shorten and accelerate their progression toward cellular senescence. This acceleration of cell senescence, i.e., the division of the remaining cells in response to cell removal (in this case, the removal of senescent cells), has predictable negative consequences.
In the case of osteoarthritis, for example, while the goal is to entirely abrogate the effects of SASP, the use of senolytics not only removes senescent chondrocytes, but risks the acceleration of senescence in the remaining cells, which were initially non-senescent, as those remaining cells must now divide to replace the missing chondrocytes (i.e., those senescent chondrocytes removed by the use of senolytic agents). This has been seen in the case of osteoarthritis [38], for example, and has been attributed to a number of possible causes [36], including the underlying causes of cell senescence and the exhaustion of stem cells, as they replace the missing senescent cells.
As we remove senescent cells from an aging tissue, local factors will signal the nearby, remaining cells to divide and replace those cells which have been removed. The subsequent cell division results in two daughter cells. Both daughter cells lose telomere length, with the result that both daughter cells are themselves that much closer to senescence. The loss of senescent cells has momentarily resulted in a mean gain in telomere length in the remaining cells (and a mean loss of cell senescence and cell dysfunction), but as the remaining cells divide to replace the missing senescent cells, there is a mean loss of telomere length in the remaining cells, with consequent acceleration in cell senescence and cell dysfunction. We should expect that the initial improvement (as measured by telomere length, cell function, SASP, etc.) will be followed by more rapid deterioration (as measured by the same criteria). Removal of senescent cells should result in a transient gain in tissue function, followed by an accelerated loss of tissue function. Senolytic therapy threatens to transiently improve clinical status, while accelerating overall age-related clinical disease.
Consider the following graphic examples. In Figure 2, telomere length is on the Y axis, while affected cells are shown on the X axis of each of the four treatment phases. In the pre- treatment phase, we see three senescent cells, whose telomere lengths indicate cell senescence, resulting in tissue dysfunction. We also see six, non-senescent cells, whose telomere lengths indicate non-senescence, thereby demonstrating normal cell function. When a senolytic treatment is applied, the immediate result is the deletion of senescent cells (“missing cells”), followed by signalling to neighboring cells (“stressed cells”), prompting the final result of cellular replication, telomere shortening, and an increased number of senescent cells. We now have six senescent daughter cells, whose telomere lengths are below the critical level and only three non- senescent cells, whose telomere lengths are above the same critical level. In this simplistic example, comparing pre-treatment to the final result, we see that we have gone from 33% (3 of 9) senescent cells to 66% (6 of 9) senescent cells, thereby accelerating the very tissue pathology that we intended to treat.

Figure 2 Senolytic effects on telomere lengths and senescence in treated tissue.
In Figure 3, we see a second graphic example in which we look down on a joint surface as composed of chondrocytes. In the pre-treatment phase, we see ten initially senescent cells are creating tissue pathology via SASP. In the intermediate phase, we have destroyed the ten senescent cells, but thereby triggered cell division in neighboring cells, whose telomeres then shorten in consequence. The final result is twenty senescent cells and an overall increase in tissue pathology via SASP. Again, we have accelerated the very tissue pathology that we intended to treat.

Figure 3 Senolytic effects on tissue senescence.
Published data (see below) is in line with the effects predicted above. Specifically, there is an initial improvement as measured by a number of bio-markers, followed by an accelerated decline in the same bio-markers. For example, in the curves shown in Figure 4, the purple curves represent mortality in untreated mice. The red curves represent mortality in mice treated with senolytics. The above theoretical considerations predict that mortality (as well as pathology) in treated animals should first show a transient improvement, which is seen in the initial flattening of the curves (here labelled as “Initial decreased mortality”, followed by a predicted acceleration of mortality (as well as pathology) in the treated animals (here labelled as “increased mortality”). These curves represent published data on senolytic therapy trials in animals [27] and should prompt concern for the outcomes in human clinical trials.
Additional adverse effects may also ensue for several other reasons. One possibility that has been raised is “the release of proinflammatory, danger-associated molecular patterns, futher exacerbating systemic chronic inflammation” [36]. Systemic side effects may also include thrombocytopenia and immune suppression [50], as well as preventing the potential roles of senescent cells in increasing stem cell differentiation, promotion of wound healing, etc. [51].
Current publications on the use of senolytic therapies suggest (short-term) benefits, but not extended benefits when followed over time, nor address long-term risks. In addition, the published data concentrates on prevention, rather than upon the effects in older organisms with established pathology, the more likely scenario in human patients. Despite these and other limitations, published studies tout senolytic therapy [28,50,52] for their clinical potential. Such studies have multiple limitations, however, including:
1) Genetically altered animals may not adequately represent the pathology found in aging human patients [16,53]. Such models frequently genetically-modify an animal (for example, to increase tau tangle production) then show that senolytics may ameliorate the artificially elevated bio-marker. Inferences to human pathology from such models are tenuous and may be both unsupported and unwarranted.
2) Studies often examine animals only through “middle age” rather than into advanced age. For example, they may follow mice to less than mid-lifespan and not into old age. As the lifespan of mice is typically 24-36 months, studies that purport to demonstrate value by following mice to only 6-8 months [16] (similar to a human of 20 years of age?) cannot adequately demonstrate effects in aging mice, let alone aging humans.
3) When animals are treated with senolytics at an appropriately advanced age, they are generally followed for only short periods, or the long-term acceleration in pathology is not mentioned in the analysis, conclusions, or discussion of the data [27].
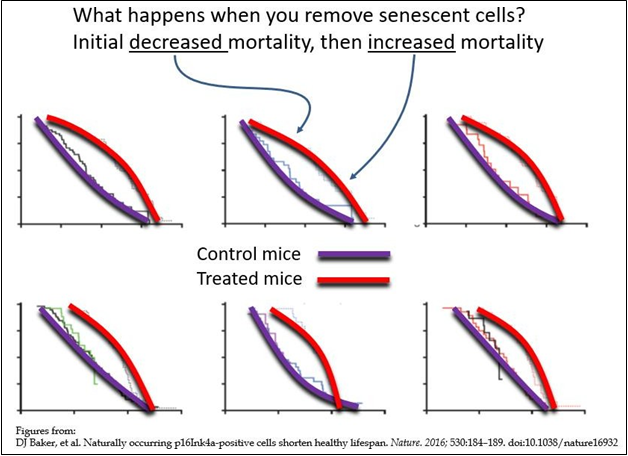
Figure 4 Senolytic agents and their outcomes.
6. Implications and Summary
Despite the lack of data on long-term human benefit or efficacy, considerable investments have been made in senolytic therapy [47,54]. The financial and the clinical risks remain underestimated. The risks are not merely those expected of any novel therapeutic agent, but are predictable, substantial, and due to naive assumptions in regard to human physiology and age- related pathology in the light of in vivo cell senescence. The acknowledged risks are routine and predictable in biotechnology: senolytics have yet to be tested in humans, there is uncertainty regarding mechanisms, and some agents are ineffective in clearing senescent cells. However, there are additional unacknowledged risks and these are substantial: senolytics will accelerate tissue pathology, exacerbate clinical disease, and cause early morbidity and mortality. Senolytic therapy will be clinically counter-productive.
The media [17,52,55,56] and companies involved in this approach remain optimistic, but the animal data suggests that any initial clinical improvement will be followed by increased pathology and an accelerated disease course. In human trials [37], we should expect acceleration of clinical disease as the outcome. Succinctly, we should rationally predict that the long-term clinical outcome with senolytics would be worse than would occur without senolytics.
Looked at in terms of the clinical lifespan of a normally aging tissue (e.g., the joint surface of a human knee as it ages and osteoarthritis ensues), we would expect function of an untreated tissue in question to decline linearly, barring clinical intervention. This normative clinical outcome can be represented (see Figure 5) as “Normal Tissue Aging” (in purple). Repetitive injury (e.g., repetitive trauma to a knee joint in the case of a basketball player, hypertension with accelerated rheological trauma in the aorta, or traumatic brain injury in the case of the CNS) can be represented as “Repetitive Injury” (in yellow). Senolytic intervention should cause an initial improvement in clinical status followed by an accelerated clinical failure with the steepening curve, represented here as “Delete Senescent Cells” (in red). This is precisely what we see in published senolytic data [27]. Finally, telomerase therapy should cause improvement at both the cell [8] and the tissue levels (as it does at organ and biomarker level [18,19]) represented here as “Reset Senescent Cells” (in green).

Figure 5 Comparison of senolytic agents, telomerase, repetitive injury, and controls.
As seen in Figure 5, the expected outcome of senolytic agents (“delete senescent cells”) contrasts markedly with the course of either untreated tissue (“normal tissue aging”) or telomerase therapy (“reset senescent cells”). Telomerase gene therapy does not accelerate senescence in the remaining cells, and results in improved tissue function and resolution of age- related disease. In contrast, senolytic agents accelerate senescence in remaining cells, and, in the long-term, would achieve the same outcome as repetitive tissue injury, i.e., accelerated tissue aging and increased clinical pathology.
The data curves of senolytic therapy display rectangularization of the disease course: the curve moves forward, followed by a more rapid acceleration of pathology. In a sense, this is a parallel to historical approaches to treating age-related disease, in which we see a rectangularization of lifespan, as we delay symptoms but do not affect the fundamental pathologic processes that underlie age-related disease.
The major risk of senolytic therapy lies not in its short-term efficacy, but in its long-term, predictably negative consequences. The short-term implications are potentially desirable; the long-term implications – as supported by a more thorough understanding of complex tissue pathology of age-related disease and by such long-term data as is available – are likely to be detrimental both to the age-related diseases for which senolytics are intended and to the lifespan of the elderly patients to whom senolytics are clinically relevant. Senolytic interventions overlook the physiology and the role of cell senescence in age-related clinical pathology and are likely to prove clinically catastrophic.
Author Contributions
The author wrote and edited the paper.
Competing Interests
The author is the founder and president of Telocyte LLC.
References
- Hayflick L, Moorhead PS. The serial cultivation of human diploid strains. Exp Cell Res. 1961; 25: 585-621. [CrossRef]
- Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965; 37: 614-636. [CrossRef]
- Harley CB, Futcher AB, Greider CW. Telomeres shorten during aging of human fibroblasts. Nature. 1990; 345: 458-460. [CrossRef]
- Allsopp RC, Vaziri H, Patterson C, Goldstein S, Younglai EV, Futcher AB, et al. Telomere length predicts replicative capacity of human fibroblasts. Proc Natl Acad Sci U S A. 1992; 89: 1011410118. [CrossRef]
- Allsopp RC, Chang E, Kashefi Aazam M, Rogaev EI, Piatyszek MA, Shay JW, et al. Telomere shortening is associated with cell division in vitro and in vivo. Exp Cell Res. 1995; 220: 194-200. [CrossRef]
- Shelton DN, Chang E, Whittier PS, Choi D, Funk WD. Microarray analysis of replicative senescence. Curr Biol. 1999; 9: 939-945. [CrossRef]
- Funk WD, Wang CK, Shelton DN, Harley CB, Pagon GD, Hoeffler WK. Telomerase expression restores dermal integrity to in vitro-aged fibroblasts in a reconstituted skin model. Exp Cell Res. 2000; 258: 270-278. [CrossRef]
- Bodnar AG, Chiu C, Frolkis M, Harley CB, Holt SE, Lichtsteiner S, et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998; 279: 349-352. [CrossRef]
- Banks DA, Fossel M. Telomeres, cancer, and aging; Altering the human lifespan. JAMA. 1997; 278: 1345-1348. [CrossRef]
- Fossel M. Telomerase and the aging cell; Implications for human health. JAMA. 1998; 279: 1732-1735. [CrossRef]
- Fossel M. Cells, aging, and human disease. New York: Oxford University Press; 2004.
- Chang E, Harley CB. Telomere length and replicative aging in human vascular tissues. Proc Nat Acad Sci USA. 1995; 92: 11190-11194. [CrossRef]
- Flanary BE, Sammons NW, Nguyen C, Walker D, Streit WJ. Evidence that aging and amyloid promote microglial cell senescence. Rejuv Res. 2007; 10: 61-74. [CrossRef]
- Boccardi V, Pelini L, Ercolani S, Ruggiero C, Mecocci P. From cellular senescence to Alzheimer's disease: The role of telomere shortening. Ageing Re Rev. 2015; 22: 1-8. [CrossRef]
- Hickman SE, Allison EK, Khoury JE. Microglial dysfunction and defective amyloid clearance pathways in aging alzheimer’s disease mice. J Neurosci. 2008; 28; 8354-8360. [CrossRef]
- Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, Baker DJ. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature. 2018; 562: 578582. [CrossRef]
- Penney J, Tsai LH. Elimination of senescent cells prevents neurodegeneration in mice. Nature. 2018; 562: 503-504. [CrossRef]
- Jaskelioff M, Muller FL, Paik JH, Thomas E, Jiang S, Adams A, et al. Telomerase reactivation reverses tissue degeneration in aged telomerase deficient mice. Nature. 2011; 469: 102-106. [CrossRef]
- de Jesus BB, Vera E, Schneeberger K, Tejera AM, Ayuso E, Bosch F, et al. Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO Mol Med. 2012; 4: 691-704. [CrossRef]
- Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM. Cellular senescence in aging primates. Science. 2006; 311: 1257. [CrossRef]
- Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010; 5: 99-118. [CrossRef]
- Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic ras and the p53 tumor suppressor. PLOS. 2008; 6: 2853-2868. [CrossRef]
- Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. 2016; 354: 472-477. [CrossRef]
- Coppe JP, Pati CK, Rodier F, Krtolica A, Beausejour CM, Parrinello S, et al. A human- like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS One. 2010; 5: e9188. [CrossRef]
- Hernandez-Segura A, de Jong TV, Melov S, Guryev V, Campisi J, Demaria M. Unmasking transcriptional heterogeneity in senescent cells. Curr Biol. 2017; 27: 2652-2660. [CrossRef]
- Freund A, Laberge R-M, Demaria M, Campisi J. Lamin B1 loss is a senescence-associated biomarker. Magin TM, ed. Mol Biol Cell. 2012; 23: 2066-2075. [CrossRef]
- Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, et al. Naturally occurring p16Ink4a- positive cells shorten healthy lifespan. Nature. 2016; 530: 184-189. [CrossRef]
- Kirkland JL, Tchkonia T, Zhu Y, Niedernhofer LJ, Robbins PD. The clinical potential of senolytic drugs. J Am Geriatr Soc. 2017; 65: 2297-2301. [CrossRef]
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011; 479: 232236. [CrossRef]
- These currently include: Unity Biotechnology, Cleara biotech, Senolytic Therapeutics, Oisin Biotechnologies.
- Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. 2016; 22: 78-83. [CrossRef]
- Grezella C, Fernandez-Rebollo E, Franzen J, Ventura Ferreira MS, Beier F, Wagner W. Effects of senolytic drugs on human mesenchymal stromal cells. Stem Cell Res Ther. 2018; 9: 108. [CrossRef]
- Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM, et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell. 2017; 169: 132-147. [CrossRef]
- Both, as of 2018, from Unity Biotechnology.
- Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, et al. The Achilles' heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. 2015; 14: 644-658. [CrossRef]
- McHugh D, Gil J. Senescence and aging: Causes, consequences, and therapeutic avenues. J Cell Biol. 2018; 217: 65-77. [CrossRef]
- Tchkonia T, Kirkland JL. Aging, cell senescence, and chronic disease - emerging therapeutic strategies. JAMA. 2018; 320: 1319-1320. [CrossRef]
- Jeon OH, Kim C, Laberge RM, Demaria M, Rathod S, Vasserot AP, et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro- regenerative environment. Nat Med. 2017; 23: 775-781. [CrossRef]
- Demaria M, O'Leary MN, Chang J, Shao L, Liu S, Alimirah F, et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 2017; 7: 165-176. [CrossRef]
- Baker DJ, Petersen RC. Cellular senescence in brain aging and neurodegenerative diseases: evidence and perspectives. J Clin Invest. 2018; 128: 1208-1216. [CrossRef]
- Kumazaki T. Modulation of gene expression during aging of human vascular endothelial cells. Hiroshima J Med Sci. 1993; 42: 97.
- Cooper LT, Cooke JP, Dzau VJ. The vasculopathy of aging. J Gerontol. 1994; 49: 191-196. [CrossRef]
- Yang Y, Wilson DL. Characterization of a life-extending mutation in age-2, a new aging gene in Caenorhabditis elegans. J Gerontol A Biol Sci Med Sci. 1999; 54: B137-B142. [CrossRef]
- Fossel M. Reversing human aging. New York: William Morrow and Company; 1996.
- Fossel M. Implications of recent work in telomeres and cell senescence. J Anti-Aging Med. 1998; 1: 39-43. [CrossRef]
- Fossel M. Cell senescence and human aging: A review of the theory. In Vivo. 2000; 14: 29-34.
- https://www.fiercebiotech.com/biotech/unity-files-for-85m-ipo-to-take-anti-aging-drugs- into-phase-1?mkt_tok=eyJpIjoiTUdJNVpUQXlZMlE1TlRObCIsInQiOiJISjNzRER4Zks1TzJTRTJ6XC9SaEVSRWJDNTMzZDFsU2Nvb0V2bHQzM2syRWdKZW1VVTJaV0xNRkFZa0JaMEZvZG43SkdNQXAzM1BobHllQURhOVhrSjJJUnFGcjlFSmQ4QWpTMGNlR2dONHhXV1RTUG5UMzkxTFBHRVhjY1JFWDMif Q%3D%3D&mrkid=20453317
- van Deursen JM. The role of senesent cells in ageing. Nature. 2014; 509: 439-446. [CrossRef]
- Shao L, Feng W, Li H, Gardner D, Luo Y, Wang Y, et al. Total body irradiation causes long-term mouse BM injury via induction of HSC premature senescence in an Ink4a- and Arfindependent manner. Blood. 2014; 123: 3105-3115. [CrossRef]
- Childs BG, Gluscevic M, Baker DJ, Laberge RM, Marquess D, Dananberg J, et al. Senescent cells: an emerging target for diseases of ageing. Nat Rev Drug Discov. 2017; 16: 718-735. [CrossRef]
- Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell. 2014; 31: 722-733. [CrossRef]
- https://www.alzforum.org/news/research-news/are-tauopathies-caused-neuronal-and-glial- senescence
- Musi N, Valentine JM, Sickora KR, Baeuerle E, Thompson CS, Shen Q, et al. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell. 2018; 17: e12840. [CrossRef]
- https://www.fiercebiotech.com/biotech/anti-aging-outfit-unity-bags-55m-and-looks-to-clinic
- https://newatlas.com/fisetin-flavonoid-anti-aging-mouse-study/56610/
- https://www.technologyreview.com/s/612284/finally-the-drug-that-keeps-you-young/
