

Mechanisms of Origin and Clinical Effects of Multiple Small Supernumerary Marker Chromosomes, Each Derived from a Different Chromosome
Ron Hochstenbach 1,* ![]() , Martin Poot 1
, Martin Poot 1![]() ,Thomas Liehr 2
,Thomas Liehr 2![]()
- Department of Genetics, University Medical Centre Utrecht, Utrecht, P.O. Box 85090, 3508 AB Utrecht,
The Netherlands - Universitätsklinikum Jena, Institut für Humangenetik, Kollegiengasse 10, D-07743 Jena, Germany
* Correspondence: Ron Hochstenbach ![]() ; Tel: +31-88-75-53800; Fax: +31-88-75-53801.
; Tel: +31-88-75-53800; Fax: +31-88-75-53801.
Received: November 15, 2016 | Accepted: January 09, 2017 | Published: February 10, 2017
OBM Genetics 2017, Volume 1, Issue 1, doi:10.21926/obm.genet.1701002
Academic Editor: Joep Geraedts
Recommended citation: Hochstenbach R, Poot M, Liehr T. Mechanisms of Origin and Clinical Effects of Multiple Small Supernumerary Marker Chromosomes, Each Derived from a Different Chromosome. OBM Genetics 2017;1(1):002; doi:10.21926/obm.genet.1701002.
© 2017 by the authors. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
Abstract
Small supernumerary marker chromosomes (sSMCs) are centric chromosome fragments additionally present in an otherwise normal human chromosome set that cannot be characterized by classical cytogenetic techniques alone. The majority of sSMCs are not yet related to a defined clinical phenotype. We compiled from the literature all 78 cases with multiple sSMCs per cell in which the chromosomal origin of the sSMCs has been identified. The number of sSMCs varies from 2 to 7; 64% have 2 sSMCs, 14% have 3 sSMCs, and the frequency decreases to 3% each for cases with 6 or 7 sSMCs. We propose that the majority of cases originate from premature separation of sister chromatids during maternal meiosis I or II, leading to multiple trisomies in the zygote. Because ~80% of single sSMCs and ~64% of sSMCs in cases with multiple sSMCs have at least one break in the centromere, we further propose that aberrant kinetochore-spindle attachment during maternal meiosis leads to breaks within or close to the centromeres of the additional chromosomes. The resulting sSMCs are mitotically stable if they contain a sufficient amount of alpha satellite repeats for proper centromere function and if the double strand breaks are repaired either by ring chromosome formation or by telomere capture or synthesis. This model is supported by observations on fertilized oocytes, polar bodies and blastomeres, which show that 2 to 7 additional chromosomes of different origin can occur in human zygotes. In addition, observations on double trisomies in cases of spontaneous abortion show that these are almost invariably of maternal origin and involve two segregation errors either during meiosis I or II, or two consecutive errors, one during meiosis I and the other during meiosis II. This model explains why all chromosomes can contribute to one of the multiple sSMCs, why each case has a unique combination of sSMCs with respect to the chromosomes of origin, why there is a maximum number of up to 6-7 sSMCs per cell, why the number of cases is inversely proportional to the number of sSMCs per cell and why all cases in which this was studied occurred de novo. We further propose that cases with a paternal origin are much rarer and result from meiotic errors that lead to sperm cells with multiple additional chromosomes. Recent studies of the parental origin of de novo multiple sSMCs in 5 patients show a maternal origin in 4 cases, involving either multiple meiosis I or II segregation errors, and a paternal origin in one case. Multiple sSMCs can lead to highly variable and complex clinical phenotypes if they contain dosage-sensitive genes. Phenotypes are further complicated by the somatic mosaicism of the sSMCs due to mitotic loss, as seen in 92% of cases. In 12.5% of postnatal and 38% of prenatal cases there are no or only very mild clinical abnormalities. Therefore, during clinical management of the pregnancy, the gene content and degree of mosaicism must be carefully evaluated for each of the multiple sSMCs.
Keywords
multiple supernumerary marker chromosomes; double trisomy; non-extrusion of polar body; premature separation of sister chromatids (PSSC); mental retardation; developmental delay
Introduction
Additional, centric chromosome fragments that are too small to be characterized unambiguously by classical cytogenetic techniques alone are termed small supernumerary marker chromosomes (sSMCs). Although some sSMCs have been associated with specific clinical disorders, for example i(12)(p10) in Pallister-Killian syndrome, i(18)(p10) in the iso-18p syndrome, and inv dup(22)(q11.2) in cat-eye syndrome, the majority of sSMCs have not yet been related to a defined clinical phenotype [1,2,3,4,5]. A single sSMC can be detected in about 0.2% of prenatal cases with ultrasound abnormalities and in about 0.3% of postnatal patients with multiple congenital anomalies and/or mental retardation (MCA/MR) [6]. Such sSMCs can be of different morphology: centric minute, ring, iso-, or inverted-duplication chromosomes have been described. Whereas most sSMCs are derived from a single chromosome, complex-rearranged sSMCs are known as well, containing material from more than one chromosome [3,7,8,9,10,11]. Several mechanisms have been proposed to explain the formation of a de novo, single sSMC, including mechanisms involving trisomic rescue [1,3,4,12] or centromere misdivision that creates a pericentromeric deletion and a compensating, ring-shaped sSMC, the so-called McClintock mechanism [13].
Patients with multiple sSMCs, that are each derived from a different chromosome, are much rarer than those with a single sSMC. Currently, there are 78 cases on record (Liehr T., 2016. Small supernumerary marker chromosomes: http://ssmc-tl.com/sSMC.html; accessed October 24th, 2016) [14]. Because previous reviews on such patients have been published more than a decade ago [15,16,17] and many additional cases have been published since, we again collected and reviewed all cases. The first purpose of this review is to discuss the possible mechanisms of origin of the multiple sSMCs. Knowing the mechanism of origin is essential for genetic counseling, since it may provide clues towards an estimate of the recurrence risk. Several models have been proposed, based on an origin from a triploid zygote [15,16,18]. However, these models were not supported by molecular investigation of the parental origin of the sSMCs. More recently, such studies were performed in selected patients, using highly polymorphic short tandem repeat (STR) markers or single nucleotide polymorphisms (SNP) [19,20,21,22]. These studies demonstrated that the multiple sSMCs either have a maternal or a paternal origin, and that in cases with a maternal origin either meiosis I or meiosis II errors are involved. Here, we discuss the extent in which the different models proposed earlier [15,16,18] and more recently [20,22] are able to explain the origin of patients with multiple sSMCs. The second purpose of this review is to provide the general characteristics of these rare patients, such as an estimate of the frequency at which they can be found among patients with multiple congenital anomalies and/or mental retardation (MCA/MR), in how many cases the multiple sSMCs originated de novo, and how many show chromosomal mosaicism. The third purpose is to assess the impact on the phenotype. For example, it is unknown how many patients do not show clinical abnormalities. This also is essential information for genetic counseling, especially when a case with multiple sSMCs has been detected during prenatal chromosome investigation.
Methods
To compile all cases with multiple sSMCs we used the terms ‘supernumerary chromosome’, ‘supernumerary marker’ and ‘supernumerary ring’ for searching PubMed (http://www.ncbi.nlm.nih.gov/pubmed) and scrutinized the retrieved publications for cases with multiple sSMCs. We also included the cases from previous reviews [3,15,16,17], and those from the sSMC-homepage curated by T. Liehr and co-workers (Liehr T., 2016. Small supernumerary marker chromosomes: http://ssmc-tl.com/sSMC.html; accessed October 24, 2016) [14]. Cases with insufficient evidence for the chromosomal origin of the multiple sSMCs were excluded. Also, cases in which the multiple chromosomes had arisen from a single chromosome by mitotic non-disjunction were excluded. Examples of such cases are case 3 of Viersbach et al. [23] and case 16 of Bartsch et al. [24]. The case of a mother and her daughter with an identical combination of multiple sSMCs [25], and a case of monozygotic twins with the same combination of multiple sSMCs [26] are each treated as a single entity.
General Characteristics of Cases with Multiple sSMCs
Table S1 shows a summary of cases with multiple sSMCs. In most cases the chromosomal origin of the sSMCs was identified by fluorescence in situ hybridization (FISH), using centromere-specific alpha satellite repeat probes, or by array-based Comparative Genomic Hybridization (array-CGH) or SNP-based array methods. For initial identification, array-based techniques are of limited use, because sSMCs lacking pericentromeric, unique DNA sequences will not be detected even if present in all cells of the patient. In addition, sSMCs containing unique, euchromatic DNA sequences may go undetected when present below a certain threshold. Using array-CGH, an sSMC can be detected if present in more than 15% of the cells in the sample [13,27,28]. When using SNP-arrays, the threshold for mosaicism detection depends on the origin of the sSMC, with a ~7% minimum, if the sSMC introduces a third haplotype, and ~12% if it does not [29,30]. Thus, in most cases the sSMCs were detected and identified using a combination of karyotyping, FISH and array-based techniques. In some cases alternative FISH methods [31] were applied such as centromere-specific multicolor FISH (cenM-FISH) and subcentromere-specific multicolor FISH (subcenM-FISH) [2], spectral karyotyping [17], or reverse FISH using Polymerase Chain Reaction (PCR)-amplified DNA from microdissected sSMCs as a probe [32].
We retrieved 72 cases in which all sSMCs were each found to be derived from a single chromosome (Table S1A- S1F). Of these, 21 were detected during prenatal diagnosis and 50 after birth, with insufficient information in one case. The sSMCs were detected in cultured amniocytes in most of the prenatal cases and in cultured peripheral blood lymphocytes in most of the postnatal cases. These 72 cases constitute the core dataset for this review. There were 6 additional cases in which one of the sSMCs was complex, containing material from more than one chromosome (Table S1G). Finally, in 7 cases the chromosomal origin of the sSMCs was not determined (Table S1H). The maximum number of sSMCs per metaphase was 6 in liveborn individuals and 7 in prenatal cases. The majority of cases (63.8%) had up to 2 sSMCs per metaphase, with the number of cases decreasing with increasing number of sSMCs (Table 1). Each case has a unique combination of sSMCs, and each of the 24 different chromosomes can give rise to one of the multiple sSMCs (Table S1).
When looking at the frequency at which the 24 different chromosomes occur in cases with multiple sSMCs, some striking observations can be made. First, the frequency at which each chromosome occurs is quite variable (Table S1, Figure 1). Chromosomes 13, 8, 6, X, 4, 12, 11 and 20 are the most frequent (together accounting for >50% of the sSMCs), and 14, 19, 21 and Y are the least frequent. Chromosomes 13, 8, 20, 6, 11 and 18 are overrepresented in cases with 2 sSMCs. For example, an sSMC(13) occurs in 30% and an sSMC(8) in 13% of such cases (Table S1, Figure 2). However, in cases with 3 and 4 sSMCs this is only true for chromosome 8, which occurs in 5 of 10 cases with 3 sSMCs and in 4 of 8 cases with 4 sSMCs. In cases with 5, 6 or 7 sSMCs, in contrast, an sSMC(8) does not occur at all (Table S1, Figure 2). A previous suggestion that an sSMC(6) is the most frequent sSMC involved [33] was wrong due to ascertainment bias.
In all cases in which cultured peripheral lymphocytes of the parents of the patient were studied, the multiple sSMCs appeared to originate de novo. Of the 37 postnatal cases in which the age at referral for karyotyping was documented, 7 were referred at or around birth (19%) and 7 others within the first year of life (19%). This indicates that in about 40% of cases the clinical symptoms were conspicuous enough to instigate karyotyping during the first year of life. The average age at ascertainment of the postnatal cases was 8.8 years, with the oldest patient being diagnosed at the age of 58 years. There are no indications for an association between the number of sSMCs and the age of ascertainment (Table 1). There are very limited data on the parental ages (Tables S1 and Table 1). The average maternal age was 28.5 years (based on 17 cases) and the average paternal age was 32.0 years (based on only 2 cases). Thus, there is insufficient information to determine if there is a parental age effect in the origin of patients with multiple sSMCs.
Table 1 Basic characteristics of cases with multiple sSMCs, each derived from a different chromosome.
|
Number of sSMCs per Metaphase |
Number of Cases with Chromosomal Origin Determined1 |
Cases with Proven Mosaicism2 |
Cases Detected3 |
Cases with de novo origin |
Average Ascertainment Age4 |
Average Maternal Age |
Average Paternal Age |
|
|
Prenatal |
Postnatal |
|||||||
|
up to 2 |
46 (63.8%) |
41 of 46 |
13 |
32 |
25 of 255 |
7.5 y (n=22) |
27 y (n=9) |
35 y (n=1) |
|
up to 3 |
10 (13.9%) |
10 of 10 |
4 |
6 |
9 of 9 |
5.8 y (n-4) |
35 y (n=2) |
no data |
|
up to 4 |
8 (11.1%) |
7 of 8 |
0 |
8 |
5 of 5 |
11.5 y (n=7) |
29 y (n=3) |
29 y (n=1) |
|
up to 5 |
4 (5.6%) |
4 of 4 |
2 |
2 |
2 of 2 |
1.8 y (n=2) |
28 y (n=2) |
no data |
|
up to 6 |
2 (2.8%) |
2 of 2 |
0 |
2 |
1 of 1 |
27 y (n=2) |
no data |
no data |
|
up to 7 |
2 (2.8%) |
2 of 2 |
2 |
0 |
1 of 1 |
prenatal |
29 y (n=1) |
no data |
|
Total |
72 |
66 of 72 (91.7%) |
21 (29.6%) |
50 (70.4%) |
43 of 43 |
8.8 y |
28.5 y |
32.0 y |
Notes: 1 cases in which the chromosomal origin of the sSMCs was unambiguously identified by FISH, array-based methods or a combination of both; 2 non-mosaic cases are defined as those in which all cells investigated contain the maximum number of sSMCs; 3 there were 71 cases for which this was documented; 4 for postnatal cases only; the two cases with up to 7 sSMCs were both prenatal cases; 5 in one prenatal case (mult 2-17, see http://ssmc-tl.com/sSMC.html) with inv dup(15) and r(X), the father had a sSMC in 2 of 100 metaphases investigated from blood, but it was not proven that this sSMC corresponded to one of those in the offspring [14].
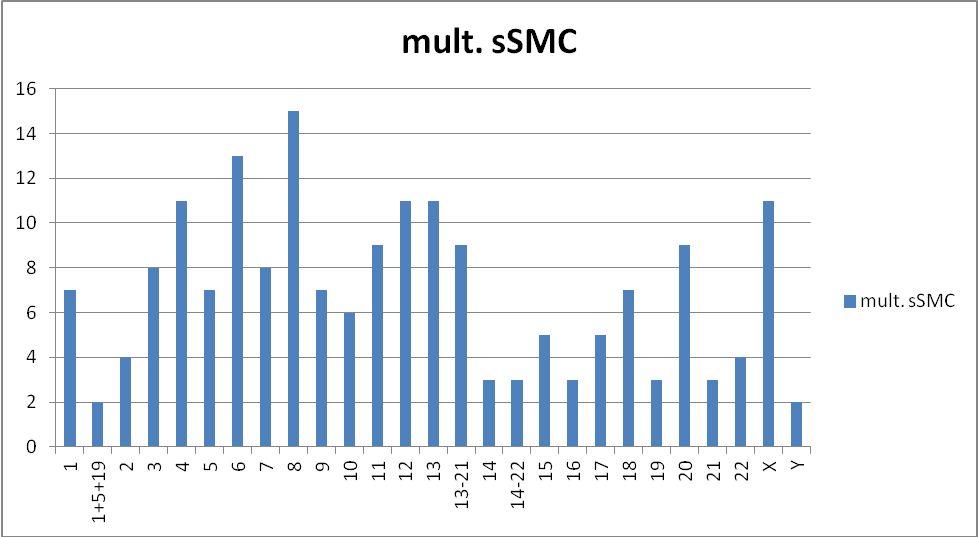
Figure 1 Frequency distribution of the 24 different chromosomes among the 72 cases with multiple sSMCs listed in Table S1A-S1F.
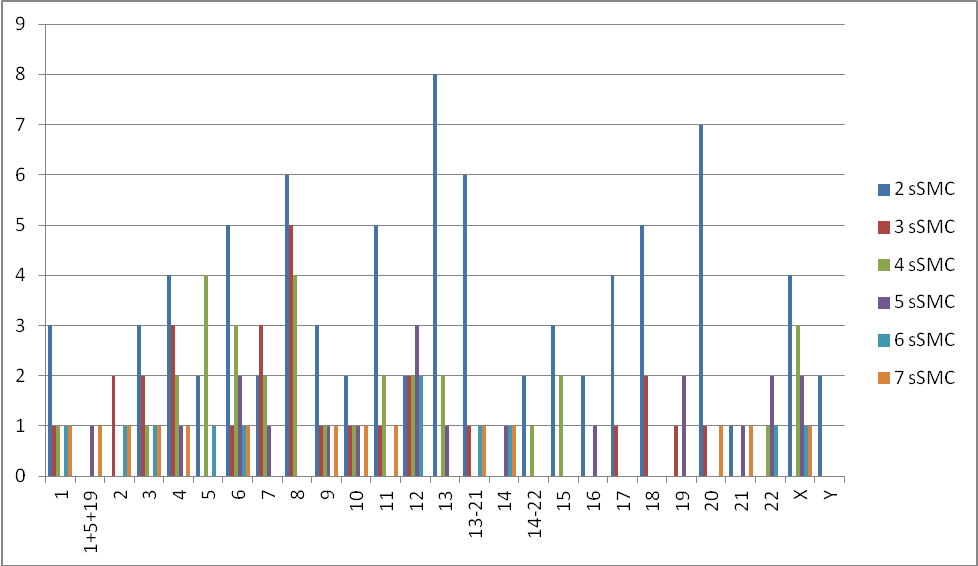
Figure 2 Frequency distribution of the 24 different chromosomes in the cases with 2, 3, 4 ,5 6, and 7 sSMCs listed in Table S1A-SiF. Because in several studies the probes used for identification of sSMCs by FISH cross-hybridized to other chromosomes, we had to include separate categories for chromosomes 1-5-19, 13-21 and 14-22.
Models for the Origin of Multiple sSMCs
Models that explain the origin of multiple sSMCs must account both for the simultaneous presence of several supernumerary chromosomes and for their small size, with chromosome breaks within or very close to the centromere. Such models must be compatible with the following observations made from the cases described in Table S1: (i) all of the 24 different chromosomes can give rise to one of the multiple sSMCs, (ii) the number of sSMCs per metaphase vary from 2 to 7, (iii) each patient has a unique combination of multiple sSMCs with respect to the chromosome of origin, (iv) the number of cases decreases with increasing number of sSMCs, (v) the multiple sSMCs occur de novo.
Initial models [15,16] were based on the assumption that a complete, third haploid chromosome complement was present in the fertilized oocyte, generating a tripronucleid embryo. Triploid embryos occur at a natural frequency of 1-3% [34,35,36]. An incomplete degradation of the third complement, irrespective of its parental origin, would produce chromosome fragments, and fragments containing a centromere would be mitotically stable and able to evolve into multiple sSMCs that were each derived from a different chromosome. In this model, cases with a paternal origin would result from the fertilization of an oocyte with two separate sperm cells [15]. Cases with a maternal origin would result from the nonextrusion of the second polar body during meiosis II, as DNA marker studies of first trimester triploid abortions show that about 70% are digynic, with errors in meiosis II in about two-thirds of cases [35,36]. In addition, as shown by direct observation, most tripronuclear zygotes that are obtained at a rate of 3-6% during ICSI result from the nonextrusion of the second polar body [37,38,39,40]. These initial models are supported by rare observations during ICSI of incomplete maternal chromatid segregation when the second polar body is extruded, leading to an additional hypohaploid maternal pronucleus [40,41], and by observations on fragmented chromosomes after in vitro fertilization [39] and in tripronuclear zygotes after ICSI [42].
Models based on zygotes with three haploid complements would explain why any chromosome can give rise to one of the sSMCs, and perhaps also why in each case a unique combination of sSMCs with respect to the chromosomal origin is present. However, it is not obvious why this model would be compatible with the observed maximum of 7 sSMCs per cell. The model also fails to explain why the number of cases decreases with increasing number of sSMCs. One would rather expect the opposite. Based on recent molecular studies of chromosome segregation errors during oogenesis and spermatogenesis we propose that multiple sSMCs are of meiotic origin, either maternal or paternal, and that this is more in line with the general observations made on the published cases. In addition, an origin from meiotic segregation errors is supported by recent molecular studies in selected patients with multiple sSMCs (summarized in Table 2).
A Postzygotic Origin of Multiple sSMCs is Unlikely
The contribution of mitotic errors to embryo aneuploidy at the cleavage stage is considerable [43]. However, several observations argue against a postzygotic origin of the multiple sSMCs seen in an individual. First, the vast majority of postzygotic segregation errors during early embryo development are chromosomal losses, not gains, as shown by FISH studies [44,45] and SNP-array studies of individual blastomeres [46]. Chromosomal losses occur three to seven times more frequently than gains [47,48]. Thus, mitotic nondisjunction, generating reciprocal losses and gains, is much rarer compared to anaphase lagging. Second, the pattern of chromosomal rearrangements seen in cleavage stage embryos (i.e. imbalances of entire chromosome arms and terminal deletions and duplications) is not compatible with the production of sSMCs [49]. Third, a rare case of monozygotic twins, each with a supernumerary r(1) and r(16), further indicates that the same two sSMCs must have been present in the zygote [26]. Finally, in all cases in which the distribution of the multiple sSMCs was studied in multiple tissues (Table 2), the sSMCs were found in both blood (mesodermal origin) and skin cells (ectodermal origin), indicating that the sSMCs were formed at a developmental stage preceding the differentiation of the primitive germ layers of the embryo. When using polymorphic STR markers that are located close to the centromere for identification of the type of meiotic error leading to a trisomy or sSMC, it is formally not possible to discriminate meiosis II errors from postzygotic mitotic errors. However, based on the considerations above, we consider a meiosis II error more likely in cases as those described by Li et al. [50] in which such a distinction was not possible.
Table 2 Patients with multiple sSMCs, each derived from a different chromosome, with parental origin determined.
|
Reference |
sSMCs |
Parental Origin |
Meiosis I or II |
Evidence and Limitations |
|
Kogan et al. (2009) [19] |
min(13);min(17) |
maternal |
n.d. |
SNP microarray (Sentrix HumanHap550); both parents investigated (limitation: no discrimination between meiosis I and II) |
|
Hochstenbach |
r(11);r(12);r(?);r(X) |
maternal |
meiosis II |
inheritance of polymorphic microsatellite markers on r(11) and r(X), present in 70% and 85% of cultured lymphocytes, respectively (limitation: father not available for analysis) |
|
Schwanitz et al. (2013) [21] |
der(1);der(12);der(18) |
paternal |
n.d. |
inheritance of polymorphic microsatellite markers on der(1) and der(18), present in 34% and 54% of cultured lymphocytes, respectively, reduction of paternal heterozygosity to homozygosity |
|
Hochstenbach |
sSMC(4);sSMC(6); sSMC(9);sSMC(14); sSMC(22) |
maternal |
meiosis II |
inheritance of SNP markers on sSMC(4), present in 90% of cultured lymphocytes (limitation: the other sSMCs could not be investigated) |
|
Hochstenbach |
sSMC(4);sSMC(8); sSMC(11) |
maternal |
meiosis I |
inheritance of polymorphic microsatellite markers on sSMC(8), present in 73% of cultured lymphocytes (limitation: three alleles in approximately 1:1:1 ratio were seen for one marker only on the sSMC(8), indicating a meiosis I origin) |
A Model for Cases with an Origin during Maternal Meiosis
A maternal origin has been found in three recently described cases. In one case, the meiotic segregation of pericentric STR and SNPs was indicative of a maternal meiosis I error, and in two other cases of a maternal meiosis II error [20,22]. We propose that such a maternal origin of the multiple sSMCs is caused by different, successive segregation errors that affect several distinct chromosomes during meiosis I, II or both. This is based on studies of the meiotic segregation of entire chromosome complements by array-CGH or SNP-array using DNA from polar body I, polar body II and the zygote [48,51-54]. For example, a gain of chromosomes 9 and 18 as a result of meiosis I errors and gains of chromosomes 4, 10, 15 and 21 resulting from subsequent meiosis II errors was found in a zygote [52]. The meiosis I gain of chromosome 9 was due to premature sister-chromatid separation, that of chromosome 18 to nondisjunction. This example shows that different kinds of several, subsequent meiotic errors lead to the presence of multiple supernumerary chromosomes in the embryo, and that gains of up to 6 different supernumerary chromosomes are possible. This is consistent with the maximum number of 6-7 multiple sSMCs, each derived from a different chromosome (Table S1). Also, any chromosome can be implicated in these meiotic errors [51,52], explaining why all chromosomes can be found as one of the multiple sSMCs, and also why each patient has a unique combination of sSMCs (Table S1). In addition, there is an inverse correlation between the number of cases and the number of supernumerary chromosomes [52], just as in cases with multiple sSMCs (Tables S1 and Table 1). This inverse correlation likely reflects a reduced viability of cases with more sSMCs and may indicate that sSMCS are, in general, efficiently eliminated until only a few remain. The frequency distribution of the chromosome of origin in multiple sSMC cases (Figure 1) does not seem to reflect that of trisomic chromosomes in zygotes with multiple supernumerary chromosomes as a result of maternal segregation errors in an a IVF population of advanced age (Table 3) [52]. For example, chromosomes 21, 22, 11 and 19 occur most frequently in such zygotes whereas in multiple sSMC cases, these chromosomes (except 11) are in the lower range of the frequency spectrum (Figure 1). Such differences can probably be explained by the various selection mechanisms that operate during development in utero and early infancy.
It may be argued that a high frequency of chromosomal aneuploidy in the zygote may be due to the ovarian stimulation treatment during IVF in women of advanced age. However, at least two studies showed that in young, naturally cycling women this treatment did not significantly increase the embryo aneuploidy rate and that even embryos of unstimulated young women showed gains of whole chromosomes [55,56]. In addition, in stimulated cycles of young women (aged 25-35) similar patterns of aneuploidies are seen as in older women, albeit at a reduced rate [57].
Studies of the origin of multiple trisomies of entire chromosomes in cases of spontaneous abortion support this model because they show that similar patterns of aberrant chromosome segregation occur during maternal meiosis. Double and triple trisomies in a nonmosaic state are not uncommon in cases of spontaneous abortion. Trisomies of two different chromosomes (see Table 4 for examples) are found in about 1.1%-1.2% of cases and those of three different chromosomes (see Table 5 for examples) in about 0.05% [58-60]. Quadruple trisomies are extremely rare but have been reported in a case of spontaneous abortion [61] and in a case of confined placental mosaicism with an origin in maternal meiosis II [62]. Parental origin studies in cases of double trisomy reveal a maternal origin for all 16 cases in which the segregation of pericentromeric STR markers yielded an unambiguous result (summarized in Table 4). In 12 of these 16 cases the double trisomy resulted from either a meiosis I or II error, whereas in 4 cases two successive errors occurred, one during meiosis I and another during meiosis II. These three scenarios are all in agreement with the aberrant segregation patterns in polar bodies and zygotes observed by single-cell array-CGH [52,53]. There are additional similarities to cases with multiple sSMCs. First, all chromosomes can be involved in cases of spontaneous abortions with double trisomy [58,60,63]. Exceptions are chromosomes 1, 3 and 19. This most likely reflects an effect on the survival capabilities of the embryo by the relatively large genetic imbalance for each of these chromosomes. Second, any combination of two chromosomes seems to be possible in double trisomies, with at least 91 out of the 300 possible combinations having been reported [58,60,61,63]. Third, there is an inverse relationship between the number of cases and the number of trisomic chromosomes [58-60]. However, the frequency distribution of chromosomes in double trisomy cases is not reflected in the frequency distribution of the chromosome of origin in cases with multiple sSMCs (data not shown). For example, chromosomes 21, X Y, 16, 18, 15 and 22, which are overrepresented in cases of double trisomy [60], occur at much lower frequencies in multiple sSMC cases (Table 3, Figure 1). Again, such differences are probably caused by the various selection mechanisms that operate during fetal development and early postnatal life.
Several predictions can be made from this model. First, when more cases are studied, cases will be identified in which some of the multiple sSMCs originated from a maternal meiosis I error, and the others from a successive error during meiosis II, similar to the situation seen in selected double trisomies of maternal origin (Table 4). Second, it can be predicted that comparable, maternal age-related mechanisms are involved in generating single, double, and triple trisomies as well as multiple sSMCs. It has been known for a long time that ovarian ageing is the principal risk factor for aneuploidy in human zygotes [reviewed by 64-67]. This is also the case for the formation of a single sSMC. From a series of 143,000 prenatal chromosome investigations it appeared that the incidence of a single, de novo sSMC significantly increases in women advanced age [11]. We therefore propose that also the formation of multiple sSMCs is related to maternal age. The largest age-dependent increase in error rate is seen for misdivision of single chromatids, also caused precocious separation of sister chromatids (PSSC) [51]. PSSC was originally reported by Angell [68], based on microscopic observations, and was subsequently shown to be strongly associated with maternal age. For example, in women older than 40 years, 90% of meiosis I errors are due to PSSC [48]. Direct observations of meiotic segregation in fixed and living oocytes of both female humans and mice have shown that the loss of cohesins during the prolonged dictyate stage is a significant contributor to meiotic segregation errors as this directly weakens centromere cohesion [65,67]. Loss of the centromeric cohesion protector Sgo2 (Shugoshin 2) in ageing female mice may also enhance cohesion loss at centromeres [69]. In addition, loss of components of the spindle assembly checkpoint such as Mad2 and Aurora C [70], a higher sensitivity to disturbances in spindle structure and a reduced energy production for spindle operation by mitochondrial dysfunction may also contribute to oocyte aneuploidy [64,71]. So far, there are very limited data on the maternal ages of patients with multiple sSMCs (Table S1, Table 1) and the hypothesis of a maternal age effect on the origin of multiple sSMCs is not yet testable.
Table 3 Comparison of chromosome of origin distribution in cases with single and multiple sSMCs, multiple gains of whole chromosomes in zygotes, and double trisomies in spontaneous abortions.
|
Zygotes1 |
Double Trisomy2 |
Multiple sSMCs3 |
Single sSMC4 |
||||
|
Chromosome Number |
Chromosome Number |
Chromosome Number |
Chromosome Number |
||||
|
21 |
9 |
21 |
134 |
8 |
15 |
15 |
236 |
|
22 |
8 |
X Y |
101 |
6 |
13 |
14/22 |
185 |
|
11 |
6 |
16 |
81 |
4 |
11 |
8 |
107 |
|
19 |
5 |
18 |
77 |
12 |
11 |
1 |
89 |
|
9 |
4 |
15 |
62 |
13 |
11 |
16 |
87 |
|
13 |
4 |
22 |
54 |
X |
11 |
22 |
87 |
|
15 |
4 |
13 |
39 |
11 |
9 |
19 |
70 |
|
17 |
4 |
8 |
36 |
20 |
9 |
14 |
68 |
|
20 |
4 |
20 |
32 |
13/21 |
9 |
18 |
60 |
|
4 |
3 |
2 |
31 |
3 |
8 |
2 |
59 |
|
16 |
3 |
14 |
29 |
7 |
8 |
5 |
58 |
|
18 |
3 |
7 |
21 |
1 |
7 |
20 |
52 |
|
6 |
2 |
10 |
13 |
5 |
7 |
9 |
44 |
|
8 |
2 |
4 |
12 |
9 |
7 |
17 |
43 |
|
10 |
2 |
9 |
12 |
18 |
7 |
21 |
42 |
|
12 |
2 |
12 |
9 |
10 |
6 |
X |
42 |
|
2 |
1 |
5 |
7 |
15 |
5 |
7 |
40 |
|
3 |
1 |
3 |
5 |
17 |
5 |
12 |
37 |
|
5 |
1 |
6 |
5 |
2 |
4 |
4 |
36 |
|
14 |
1 |
11 |
5 |
22 |
4 |
3 |
34 |
|
1 |
0 |
17 |
5 |
14 |
3 |
11 |
32 |
|
7 |
0 |
1 |
0 |
16 |
3 |
Y |
31 |
|
X |
0 |
19 |
0 |
19 |
3 |
6 |
24 |
|
Y |
0 |
X |
0 |
21 |
3 |
10 |
18 |
|
1/5/19 |
0 |
1/5/19 |
- |
14/22 |
3 |
13 |
11 |
|
13/21 |
0 |
13/21 |
- |
Y |
2 |
1/5/19 |
13 |
|
14/22 |
0 |
14/22 |
- |
1/5/19 |
2 |
13/21 |
64 |
|
X Y |
0 |
Y |
0 |
X Y |
0 |
X Y |
0 |
|
Total |
69 |
|
770 |
|
186 |
|
1,669 |
Notes: 1 data from Handyside et al. (2012) [52]; numbers specify the chromosomal gains due to maternal segregation errors in zygotes with multiple gains of maternal origin (n = 30 cases); 2 data from Micale et al. (2010) [60]; numbers specify the occurrence of each chromosome in a double trisomy in cases of spontaneous abortion (n = 385 cases); 3 data from Tables S1A-S1F, including only cases in which the chromosomes of origin are known (n = 72 cases); 4 data from the sSMC database (http://ssmc-tl.com/sSMC.html, accessed October 24, 2016) [14]; in order to correct for recurrent sSMCs, cases with i(9)(p10), i(12)(p10), inv dup(15), i(18)(p10), inv dup(22) and der(22)t(11;22)(q23;q11.2) sSMCs were excluded; also complex sSMCs and sSMCs with a neocentromere were excluded, as the latter arise by breaks at a different position than predicted by the model for the origin of multiple sSMCs described in the text; sSMCs derived from the X and Y chromosomes were included only if they occurred in a karyotype with 46 structurally normal chromosomes (n = 1,669 cases).
Table 4 Parental origin of non-mosaic double trisomy determined by segregation of STR markers.
|
Study |
Karyotype |
Ascertainment1 |
Maternal Age (Years) |
Paternal Age (Years) |
Parental Origin and Meiotic Segregation Error |
|
Zaragoza et al. 1994 [144] |
48,XX, 4, 14 |
spontaneous abortion |
37 |
40 |
mat MII |
|
Zaragoza et al. 1994 [144] |
48,XX, 10, 15 |
spontaneous abortion |
44 |
43 |
mat MI or MII |
|
Zaragoza et al. 1994 [144] |
48,XY, 15, 16 |
spontaneous abortion |
36 |
34 |
mat MI or MII |
|
Zaragoza et al. 1994 [144] |
48,XY, 15, 21 |
spontaneous abortion |
42 |
50 |
mat MI |
|
Park et al. 1995[145] |
48,XXX, 21 |
amniocentesis 17.5w |
19 |
19 |
mat MII |
|
Chen et al. 2000 [146] |
48,XXX, 18 |
IUGR 34w |
26 |
n.d. |
mat MII |
|
Robinson et al. 2001 [147] |
48, 14, 21 |
spontaneous abortion |
42 |
n.d. |
pat MI or MII(14)/mat MI(21) |
|
Li et al. 2005 [50] |
48,XX, 16, 22 |
spontaneous abortion |
n.d.2 |
n.d. |
mat MI |
|
Li et al. 2005 [50] |
48,XXY, 18 |
spontaneous abortion |
n.d.2 |
n.d. |
mat MI |
|
Li et al. 2005 [50] |
48,XX, 15, 21 |
spontaneous abortion |
n.d.2 |
n.d. |
mat MII3 |
|
Li et al. 2005 [50] |
48,XX, 2, 5 |
spontaneous abortion |
n.d.2 |
n.d. |
mat MII3 |
|
Diego-Alvarez et al. 2006 [63] |
48,XX, 15, 22 |
spontaneous abortion 7w |
37 |
39 |
mat MI |
|
Diego-Alvarez et al. 2006 [63] |
48,XX, 8, 21 |
spontaneous abortion 8w |
40 |
49 |
mat MI |
|
Diego-Alvarez et al. 2006 [63] |
48,XXX, 18 |
spontaneous abortion 13w |
37 |
43 |
mat MII |
|
Diego-Alvarez et al. 2006 [63] |
48,XY, 18, 22 |
spontaneous abortion 8w |
43 |
60 |
mat MII(18) / mat MI(22) |
|
Biselli et al. 2009 [148] |
48,XXY, 21 |
birth of liveborn baby |
13 |
n.d. |
mat MI |
|
Guzel et al. 2009 [149] |
48,XXX, 21 |
abnormal ultrasound findings |
n.d. |
n.d. |
mat MII(X) / mat MI(21) |
|
Chen et al. 2011 [150] |
48,XXY, 18 |
amniocentesis 18 w |
42 |
43 |
mat MII(X) / mat MI(18) |
|
Sheth et al. 2011 [151] |
48,XXX, 21 |
10-days old baby |
26 |
29 |
mat MI(X) / mat MII(21) |
Notes: 1 IUGR = intra uterine growth retardation; 2 The mean maternal age in the study of Li et al. 2005 [50] was 34.5 ± 6.6 years; 3 Li et al. 2005 [50] interpreted the marker segregation in the 48,XX, 15, 21 and 48,XX, 2, 5 cases as indicative of postzygotic mitotic nondisjunction of the maternal chromosome. However, a meiosis II error is more likely (see text).
Table 5 Cases of nonmosaic triple and quadruple trisomy in spontaneous abortion1.
|
Study |
Karyotype |
Tissue Studied2 |
Gestation Age (Weeks) |
Maternal Age (Years) |
|
Kajii et al. 1980 [152] |
49,XX, 2, 5, 8 |
sac and chorionic villi |
6 |
n.d. |
|
Hassold et al. 1984 [153] |
49,XX, 14, 15, 22 |
n.d. |
8 |
42 |
|
Chavarro et al. 1990 [154] |
49,XY, 8, 13, 18 |
placenta |
10 |
29 |
|
Johnson et al. 1990 [155] |
49,XX, 18, 21, 22 |
chorionic villi |
11 |
40 |
|
Petrella et al. 1991 [156] |
49,XX, 2, 8, 11 |
chorionic villi |
9 |
33 |
|
Pettenati and Rao 1991 [157] |
49,XXX, 5, 13 (fetus mosaic with 10% 46,XX) |
placenta and fetus |
17 |
37 |
|
Soukup 1992 [158] |
49,XX, 5, 16, 20 |
sac and chorionic villi |
12 |
34 |
|
Reddy et al. 1999 [59] |
49,XXX, 18, 21 |
sac and chorionic villi |
8 |
34 |
|
Reddy et al. 1999 [59] |
49,XXY, 15, 20 |
chorionic villi |
18 |
38 |
|
Reddy et al. 1999 [59] |
49,XX, 2, 8, 12 |
chorionic villi |
7 |
33 |
|
Reddy et al. 1999 [59] |
49,XY, 20, 21, 22 |
chorionic villi |
6-8 |
46 |
|
Subramaniyam et al. 2014 [61] |
49,XX, 14, 21, 22/47,XX, 22 |
n.d., missed abortion after IVF |
n.d. |
41 |
|
Subramaniyam et al. 2014 [61] |
50,XX, 8, 16, 20, 22/49,XX, 16, 20, 22 |
n.d., missed abortion after IVF |
7 |
41 |
|
Subramaniyam et al. 2014 [61] |
49,XX, 8, 20, 22/47,XX, 22 |
n.d. |
8 |
41 |
|
Subramaniyam et al. 2014 [61] |
49,XXX, 5, 8 |
n.d., missed abortion after IVF |
8 |
38 |
Notes: 1 The table only shows cases in which there was no evidence for confined placental mosaicism of the aneuploid cells; 2 the materials studied were the products of conception; in most studies this was limited to chorionic villi.
Possibly, also variation in recombination rate influences the frequency at which trisomies arise in the oocyte. There is extensive interindividual variation in the number and distribution of recombination events on the 22 autosomes, in both males and females, as shown by linkage analysis and SNP-based genotyping in pedigrees from several families [72-74]. As determined by the number of MLH1 foci in oocytes, there is a ~10-fold difference in crossover numbers between normal females [66]. Lack or reduced levels of recombination have been found to be associated with trisomies of chromosomes 15, 16, 18, 21 and X [75]. The risk for PSSC increases in non-recombinant chromatids as shown by SNP-based genotyping of meiotic products [54]. Therefore, it is possible that the mothers of cases with multiple trisomies or sSMCs may be at the extreme lower end of the spectrum of normal variation in the number of crossovers per oocyte.
An alternative possibility for a maternal origin of multiple trisomies or sSMCs is gonadal mosaicism that arises by segregation errors during the pre-meiotic divisions in the developing ovary. This possibility must be considered because such cases cannot be discriminated from a maternal meiosis I error when using pericentromeric DNA-markers. There is robust evidence for the existence of mosaicism with trisomic primordial germ cells in the ovary, not in the testes [76]). In addition, examples have been described of oocytes that were never exposed to sperm but that contained multiple, additional chromosomes that originated by segregation errors during the mitotic divisions of the primordial germ cells [77]. However, based on an estimated prevalence of ~5% in young couples with a child with Down syndrome [78], gonadal mosaicism would provide an explanation in only a subset of cases with multiple trisomies or sSMCs, and probably only in women of relatively young age. This subset would shrink rapidly with advancing maternal age, as there is no evidence that pre-meiotic segregation errors of chromosomes 13, 16 or 21 can explain the age-related increase in the rate of the associated trisomy [79].
A Model for Cases with an Origin During Paternal Meiosis
A paternal origin for cases with multiple sSMCs as a consequence of a double fertilization event was initially proposed by Beverstock et al. [15]. In an attempt to find a unified model for the origin of triploidy, twinning, mozaicism, chimaerism and trisomy, Golubovsky [18] argued that fertilization by two separate sperm cells introduces two pairs of paternal centrosomes in the zygote, leading, in most cases, to a tripolar spindle. This would result in chaotic chromatid segregation and gross aneuploidy with, in rare cases, multiple trisomies of paternal origin as a possible outcome. Survival to term would depend on the fragmentation of the supernumerary chromosomes to multiple sSMCs. In theory, this could be an explanation for a paternal origin.
Based on the observation that all males produce low levels of sperm cells with disomy of a single chromosome, as shown by FISH [80], we propose that the fertilization by a single sperm cell that carries several additional chromosomes would explain cases with a paternal origin of the multiple sSMCs. The highest mean rate of disomy in spermatozoa of normal, healthy males has been found for chromosome 22 (0.47%), the lowest for chromosome 8 (0.03%) [80]. Assuming independent events, multiple additional chromosomes would be possible as well, but a lower rate. For example, there is at least one case of a triple trisomy of paternal origin, involving chromosomes 7, 13, and 20, that most likely originated from a meiosis II error [81]. Such a triple trisomy may be the explanation for the case of a patient with a supernumerary sSMC(1), sSMC(12) and sSMC(18) [21]. A triple trisomy for chromosomes 1, 12 and 18, with sperm disomy rates of 0.08%, 0.14% and 0.06%, respectively [80], would be expected at a rate of 0.0008 x 0.0014 x 0.0006 = 0.000000067% of sperm cells. Overall, about 2.5% of spermatozoa from healthy, normal males contain one additional chromosome [80]. Again assuming independent events, about 0.0625% would carry two additional chromosomes, and about 0.0016% three. Based on such low frequencies, cases of paternal origin would be very rare. Also parental origin studies of double trisomies in cases of spontaneous abortion, which failed to demonstrate a paternal origin in 16 cases investigated (Table 4), indicate that paternal cases will be vastly outnumbered by maternal ones. In addition, the sperm disomy rates of the 24 different chromosomes [80] is not reflected in their frequency of occurrence among the 46 cases with two sSMCs (Table S1). This, also, does not support a paternal origin for most cases. The fathers of rare, paternally derived cases may be exceptional males, having much higher sperm disomy frequencies compared to the average male. Indeed, some (healthy) males display elevated sperm disomy rates for specific chromosomes [82,83] and other males for all chromosomes, such as the father of four consecutive trisomic pregnancies who had 5-6 times elevated sperm disomy rates for all chromosomes tested [84]. Conceivably, such males have a low number of crossovers per spermatocyte, as indicated by the finding that two-thirds of 47,XXY males of paternal origin resulted from the achiasmatic nondisjunction of the X and Y chromosomes [85].
The Majority of sSMCs Have a Break in the Centromere
Irrespective of their paternal origin, the supernumerary, entire chromosomes must have been converted into much smaller sSMCs. Insights in the underlying mechanism can be provided by precisely identifying the location of the breaks in the sSMCs. This has been achieved by the development of bacterial artificial chromosome (BAC) probes for FISH or array-CGH that represent the proximal-most, unique DNA sequences for each chromosome arm [13,86-88]. Following this approach, it could be shown that approximately 82% of nonacrocentric, ring-shaped sSMCs have one or both breaks within the centromeric alpha satellite repeat array, with euchromatic DNA from only one arm being detectable [13]. The other 18% of sSMCs have breaks in both the short and the long arm, each within a few Mb from the centromere. This indicates that in the large majority of single sSMCs, one of the breaks is located within the centromere.
Mapping studies of breakpoints in patients with multiple sSMCs are in agreement with these findings for single sSMCs. For example, detailed molecular mapping of an sSMC(12), detected in a mentally retarded male who had up to 6 sSMCs per metaphase, showed the absence of all tested STR markers and P1-derived artificial chromosome (PAC) and yeast artificial chromosome (YAC) clones from the p-arm, indicating that one break was in the centromeric alpha satellite repeats [32]. From the cases described in Table S1, mapping data based on either FISH or array investigations (see http://ssmc-tl.com/sSMC.html) were available for 107 sSMCs (excluding recurrent inv dup(15), and irrespective of being ring-shaped or not). Of these, 36.5% contained DNA sequences from both chromosome arms, 36.5% from one arm only, and 27.0% were centric fragments that did not carry unique pericentromeric DNA sequences. Thus, also in patients with multiple sSMCs, the majority (64%) of the sSMCs have at least one break within the centromere, similar to the situation in patients with a single sSMC [13].
Based on this observation, we propose that maternal centromere misdivision represents the major pathway for sSMC formation from an intact, ancestral chromosome, both in cases with a single sSMC [13] and with multiple sSMCs. This is supported by several other observations. If misdivision of the maternal centromere is the major pathway for sSMC formation, maternal UPD for the homologous, structurally normal chromosome will not occur. In cases with multiple sSMCs where this has been studied, this is indeed the case [12,20,22]. Furthermore, during tumorigenesis [89] and in cell lines with defective spindles [90,91], breaks in or near centromeres have been observed when kinetochores attach to both spindle poles (so-called merotelic attachment). The resulting opposite pulling forces exerted by the microtubules on the kinetochore lead to DNA double strand breaks (DSBs) that are within or in close proximity to the centromere. Another contributing factor may be a crossover event very close to a centromere, as this is known to contribute to meiotic segregation errors [75]. Normally, recombination in centromeres is suppressed, possibly by an absence of DSB formation or DSB repair [92]. Recombination close to a centromere is related to defective segregation during meiosis I and to premature separation of sister chromatids in the yeast Saccharomyces cerevisiae [92]. In humans, a significant proportion of trisomy-X of maternal origin had a recombination event close to the centromere [93,94] and it has been shown for chromosome 21 that pericentromeric crossovers occur more frequently in ageing women [95]. It has been proposed that recombination close to a centromere disturbs cohesion in the pericentric region or proper kinetochore orientation to the spindle poles, leading to merotelic attachments with (peri)centric DSB as a consequence [92]. Although for unknown reasons the extent of suppression of centromeric recombination can be very variable, even between oocytes of the same donor [54], this may explain why some oocytes can have multiple, supernumerary chromosomes.
As a next step in sSMC formation, the centromere-associated DSBs are processed in several ways to yield the different types of sSMCs that we see in the patients listed in Table S1. The DSB in or close to the centromere may activate DNA damage repair, either leading to small ring-shaped sSMCs by the joining of both ends, or to centric, minute and linear sSMCs by recombination-mediated telomere capture or de novo TTAGGG repeat synthesis. An sSMC is mitotically stable if the amount of centromeric alpha satellite repeat sequences retained in the sSMC is sufficiently large. For example, an sSMC(17) found in a mother and her two children and containing only 20-30% of the normal amount of alpha satellite repeats (~700 kb) has a fully functional centromere [96]. In a case with an sSMC(5) and sSMC(6) we found that the alpha satellite repeat array was so much reduced in the sSMC(5) that it was undetectable by FISH using DZ52 as a probe, being detectable only after microdissection and reverse FISH [33]. Such cases suggest that the centromeric alpha satellite repeat array in an sSMC can be much smaller than in the chromosome of origin. Indeed, studies on artificial chromosomes in cultured human cells reveal that a minimum of ~100 kb of alpha satellite repeats is required for mitotic stability [97]. This may also be the case in sSMCs. The size of the centromeric alpha satellite repeat array varies from ~300 to ~5,000 kb in the different human chromosomes, with a 2-4 fold size polymorphism among homologous chromosomes [98,99], but there are no indications that longer arrays are associated with a higher frequency of the corresponding sSMCs in multiple sSMC cases.
This model for the origin of multiple sSMCs is supported by the finding that, in mouse oocytes, experimentally induced DSBs delay the extrusion of the first polar body due to a disrupted attachment of microtubules to kinetochores and activation of the spindle assembly checkpoint [100,101]. In metaphase spreads at meiosis I and meiosis II of such oocytes fragmented chromosomes and misaligned chromosome fragments were observed [100,101]. This suggests that the formation of the sSMCs takes place during the final stages of oocyte maturation (meiosis I) and/or around fertilization (meiosis II), within the female pronucleus. Some chromosome fragments could evolve into multiple, mitotically stable sSMCs. We further propose that the overwhelming majority of the sSMCs evolves from a pericentric chromosome fragment that is generated by two breaks located within or close to the centromere, and, hence, contains material from one chromosome of origin. Only in six exceptional cases (summarized in Table S1G), one of the multiple sSMCs has been shown to be complex, containing material from two or three distinct chromosomes. In none of these cases the breakpoints of the chromosomal segments involved have been determined at nucleotide resolution and, therefore, the possible mechanisms that generated these complex sSMCs, such as non-allelic homologous recombination (NAHR), fork stalling and template switching (FoStTeS), or microhomology-mediated break-induced repair (MMBIR) [102], have not been ascertained. However, as is true for the majority of sSMCs, in at least three of these complex sSMCs one of the contributing chromosome fragments was also generated by a break within the centromere [103,104,105].
A General Scenario for the Origin of Multiple sSMCs
We propose that the first step in the formation of multiple sSMCs is the occurrence of multiple segregation errors during maternal meiosis I, II or both. As shown by studies in oocytes and polar bodies, this can generate up to 6 or 7 supernumerary, complete chromosomes in the zygote. The finding that ~82% of single sSMCs and ~64% of multiple sSMCs have at least one break within the centromere is suggestive of a defective arrangement of the kinetochores of the chromosomes involved during meiosis, as seen in merotelic attachments in tumor cells that lead to centromere instability. Such events are associated with maternal age, with loss of centromeric cohesins as an important contributing factor, perhaps together with ectopic DSBs or recombination within or close to the centromere. Thus, within the female pronucleus, multiple sSMCs originate from centric fragments containing variable amounts of pericentric euchromatin. As a second step, the breaks are repaired by telomere capture or de novo TTAGGG synthesis, generating centric minute sSMCs, or, alternatively, by ring formation. If the amount of alpha repeats is sufficiently large, the sSMCs will be mitotically stable and persist throughout the cleavage divisions of the embryo. This model explains why multiple sSMCs occur as de novo, sporadic events, why all chromosomes may contribute to the multiple sSMCs, why any combination of chromosomes of origin seems to be possible, why there is a maximum of 6-7 sSMCs per cell, and why the number of cases is inversely proportional to the number of sSMCs per cell. We propose that cases of paternal origin are much rarer and find their explanation in the fertilization of an euploid oocyte by a sperm that carries multiple, supernumerary chromosomes. Thus, several different mechanisms are involved in generating these extremely rare karyotypes, consistent with several recent studies (summarized in Table 2) in which the parental origin of at least one of the sSMCs could be identified.
Frequent Somatic Mosaicism Associated with Multiple sSMCs
Recently, long-term in vitro studies [106,107] showed that sSMCs are unstable during mitosis, as is the case also in vivo in sSMC carriers, with dicentric inverted duplication type sSMCs being more stable compared to monocentric rod- or ring-shaped sSMCs [107]. This may reflect a reduced centromere function in monocentric sSMCs, as these are frequently generated by breaks within the centromere, leading to loss of alpa satellite repeats. It is not unexpected, therefore, that somatic mosaicism is a frequent feature in cases with multiple sSMCs (Table S1, Table 1), as it is in cases with a single sSMC [108]. Of the 72 cases, 66 showed mosaicism (91.7%). Chromosomal mosaicism was considered to be proven if there were different numbers or combinations of sSMCs per cell. We considered mosaicism to be absent if all cells investigated contained the maximum number of sSMCs. In 9 of the 66 cases with mosaicism a second cell type was investigated (usually these were cultured skin fibroblasts in postnatal cases and peripheral blood in most prenatal cases) and in all of these the mosaicism was documented for the other cell type as well. In 8 of these 9 cases the degree of mosaicism was comparable between the two tissues investigated, except for one case with a supernumerary r(7) and min(13) sSMC in which the percentage of metaphases with sSMCs varied from 11% in skin fibroblasts to 100% in blood (Table S1, case mult 2-26). Indeed, it is well established that the percentage of cells containing a single sSMC can vary considerably between different tissues [108]. Of the 21 prenatal cases with multiple sSMCs there were only 3 without apparent mosaicism, indicating that in most cases the mitotic loss of the sSMCs started before birth.
Assuming that the multiple sSMCs are present in the zygote (as found in a unique case of monozygotic twins, described by Shanske et al. [26]), it can be concluded that mitotic loss during subsequent development of the embryo leads to chromosomal mosaicism in more than 90% of individuals with multiple sSMCs. Perhaps chromosomal mosaicism could be detected in all individuals with multiple sSMCs, provided that sufficient cells from several tissues are studied. As shown by some of the 66 cases with mosaicism (Table S1), this leads to divergent numbers and combinations of the multiple sSMCs in different cell types. Such complex patterns of chromosomal mosaicism complicate genotype-phenotype correlations [108] because each of the multiple sSMCs may have its own idiosyncratic pattern of distribution in the different tissues and organs of the patient.
Estimating the Frequency of Individuals Carrying Multiple sSMCs
The frequency of individuals in the general population with multiple sSMCs is unknown. To get some insight into their prevalence we compiled surveys of large population cohorts, both prenatal and postnatal, in which more than 10,000 individual cases were included (Table 6, also see [6] for studies based on smaller cohorts). All of these studies were based on karyotyping of unselected, consecutive pregnancies or unselected postnatal cases (liveborn infants or healthy, normal sperm donors). In unselected prenatal cases, a single sSMC was found at a frequency of 0.067%, while in postnatal cases this was 0.039%. These numbers are in line with those from a review of 132 studies, comprising 1,288,693 cases, in which single sSMCs were found in 0.075% of prenatal cases, and in 0.044% of postnatal cases [6]. Assuming that cases with multiple sSMCs would also have been detected in the studies listed in Table 6, it is apparent that cases with multiple sSMCs are much rarer compared to those with a single sSMC. This suggests that the presence of multiple sSMCs has a severe negative effect on viability. This is particularly striking for the postnatal cases, in which not a single case with multiple sSMCs was detected among 116,899 unselected individuals. In unselected prenatal investigations the frequency can be estimate at 5.7 to 11.3 per million.
Table 6 Incidence of cases with multiple sSMCs in series of unselected cytogenetic investigations, each comprising more than 10,000 cases.
|
Authors [Reference] |
Year |
City or Company, Country |
Cohort Type |
Number of Cases Investigated |
Cases with one sSMC |
Cases with Multiple sSMCs, Each Originating from a Different Chromosome |
|
|
Hook and Cross [140] |
1987 |
New York, USA |
prenatal |
75,924 |
60 |
2 possible cases 1 |
|
|
Warburton [159] |
1991 |
several cities, USA |
prenatal |
377,357 |
164 |
2 possible cases2 |
|
|
Jacobs et al. [160] |
1992 |
Salisbury, UK |
prenatal; maternal age >35 yrs |
14,677 |
23 |
0 |
|
|
Blennow et al. [161] |
1994 |
several cities, Sweden |
prenatal |
39,105 |
31 |
0 |
|
|
Bartsch et al. [24] |
2005 |
Düsseldorf, Germany |
prenatal |
43,273 |
42 |
03 |
|
|
Huang et al. [126] |
2006 |
Genzyme, California, USA |
prenatal |
100,000 |
100 |
5 proven + 1 possible case4 |
|
|
Forabosco et al. [162] |
2009 |
98 laboratories, Italy |
prenatal; maternal age <35 yrs |
37,207 |
23 |
0 |
|
|
Forabosco et al. [162] |
2009 |
98 laboratories, Italy |
prenatal; maternal age >35 yrs |
51,758 |
45 |
0 |
|
|
Malvestiti et al. [11] |
2014 |
Busto Arsizio, Italy |
prenatal |
143,000 |
104 |
0 |
|
|
Total |
882,301 |
592 |
5 proven + 5 possible cases |
||||
|
Hook and Hamerton [163] |
1977 |
Canada, Denmark, UK, USA5 |
postnatal; live born infants |
56,952 |
10 |
0 |
|
|
Nielsen and Wohlert [164] |
1991 |
Århus, Denmark |
postnatal; live born infants |
34,910 |
24 |
0 |
|
|
Maeda et al. [165] |
1991 |
Sagamihara, Japan |
postnatal, live born infants |
14,835 |
4 |
0 |
|
|
Ravel et al. [166] |
2006 |
France |
postnatal, healthy sperm donors |
10,202 |
4 |
0 |
|
|
Total |
116,899 |
42 |
0 |
||||
Notes: 1 These are case BDI 40083 with 1-3 sSMCs per metaphase and case case BDI 41926 with 3-5 sSMCs per metaphase. However, in these two cases there is no evidence that the sSMCs originated from different chromosomes: 2 These are mosaic cases with “a cell line with two or three copies of a marker chromosome”; there is no evidence that these sSMCs originated from different chromosomes; 3 Bartsch et al. 2005 [24] describe three cases with mosaicism for 1-2 sSMCs, with the sSMCs presumably derived from the same chromosome in each case: 4 Huang et al. 2006 [126] identified five cases (see Table 1) in which 2 to 4 sSMCs were detected that were each derived from a different chromosome (cases 103, 104, 105, 106 107); they also describe a case with two different sSMCs without evidence for an origin from different chromosomes (case 108); 5 based on several published series originating from Århus (Denmark), Winnipeg (Canada), Edinburgh (UK), London (UK), Boston (USA), New Haven (USA).
Some assumptions must be made to derive an estimate for the frequency of multiple sSMC cases in postnatal cases with MCA/MR. In such patients the frequency of a single sSMC is 0.288%, based on 69,332 cases from 26 studies [6]. A comparable estimate for multiple sSMCs among MCA/MR patients is not available but is likely to be much lower. A first estimate can be based on the finding that in unselected prenatal investigations, cases with multiple sSMCs are about 59 -118 times less frequent than those with a single sSMC (see Table 6). If it is assumed that the same is true among MCA/MR patients, the rate in this group would be 0.0024% - 0.0049% MCA/MR patients (equivalent to one case in 20,000 - 40,000 cases). A second estimate is based on the fact that about 1.9% of all the 3,986 sSMC cases listed in the sSMC Database, irrespective of the phenotype, are cases with multiple sSMCs (http://ssmc-tl.com/sSMC.html) (this is somewhat higher than the 1.4% reported in 2004, based on 1,398 cases in the database at that time [3], probably reflecting a biased reporting of the unusual cases with multiple sSMCs during the last decade). If it is assumed that also 1.9% of MCA/MR patients with sSMCs have multiple sSMCs, the rate would be 1 in 18,300 cases. This fits well with the lower estimate of 1 in 20,000 derived above. We conclude that in MCA/MR patients, the rate of cases with multiple sSMCs is between one in 20,000 to 40,000. This estimate is compatible with a survey of diagnostic kayotyping in the Netherlands [109], with one case [15] detected among all of the 36,325 consecutive MCA/MR referrals during a 10-year period.
Clinically Abnormal Phenotypes in Patients with Multiple sSMCs
A first step in a systematic approach to predict the phenotype in cases with multiple sSMCs is to precisely determine the size and the gene content of each of the sSMCs. A second step is to determine, if possible, the distribution and degree of mosaicism of each sSMC in various tissues of the patient. Of course, an sSMC that only contains repetitive DNA-sequences, such as centromeric alpha repeats or pericentromeric satellite repeats, will be innocuous (provided that there is no uniparental disomy (UPD) of the structurally normal, homologous chromosomes).
The phenotypes of the individuals with multiple sSMCs listed in Table S1 are highly variable. This makes it on the one hand difficult to attribute specific phenotypes to a given sSMC. On the other hand, due to its size, an sSMC contains only a limited number of plausible candidate genes. A phenotypic effect can be expected if the sSMC extends beyond the so-called “uncritical region”, which is defined as the maximum pericentromeric region that does not produce a phenotype when present in three copies per cell. These regions have been defined for each chromosome, based on normal individuals that carry either a single sSMC or a duplication of a pericentromeric segment (see the sSMC database at (http://ssmc-tl.com/sSMC.html). Taking into account that an sSMC contains an additional copy of a chromosomal segment one has to assume that its genes exert their phenotypic effects via a dosage dependent mechanism, further limiting the number of candidate genes [102,110]. An essential second step is to determine the level of mosaicism of each of the sSMCs, if possible in multiple tissues. When the chromosomal origin of each sSMC is known, targeted FISH investigations using centromere-specific DNA probes can be instigated on interphase cells from buccal mucosa and urine sediment, for example. An SMC with an effect on the phenotype is expected to be present in the majority of cells investigated.
An example of this approach is provided by the case of a 24-year old male with mild intellectual disability and multiple congenital heart defects who carried a supernumerary r(11), r(12) and r(X) in both blood and skin cells [20]. The r(11) and r(12) sSMCs did not extend beyond the uncritical region of the corresponding chromosomes and were present in ~50% to ~70% of cells. Therefore, they were considered to be of minor relevance for the phenotype. In contrast, the r(X) sSMC extended beyond the uncritical region, it was genetically active as it lacked the XIST gene, it was present in ~90% of cells examined and contained dosage-sensitive candidate genes for intellectual disability (ARHGEF9) and heart defects (FAM123B). ARHGEF9 encodes a protein called collybistin that binds in a 1:1 stoichiometry to gephyrin trimers during the microtubule-mediated transport of trimers to the postsynaptic plasma membrane. Here, the gephyrin trimers form a hexagonal lattice that serves to cluster specific receptors at inhibitory post-synaptic sites in the brain [111,112]. It was proposed that the two-fold increase in ARHGEF9 copy number leads to an excess amount of free collybistin, leading to blocking of the anchoring points for gephyrin lattice assembly, and ultimately, contributing to the intellectual disability of the patient. FAM123B encodes a component of the β-catenin degradation complex that inhibits WNT signaling. Because perturbation of WNT signaling is associated with heart defects in animal model systems, the excess copy of FAM123B may contribute to the cardiac phenotype of the patient [20].
Such a systematic analysis of the genotype-phenotype relationship has not been possible in most cases due to a lack of precise delineation of the gene content in the sSMCs. Of the 72 cases summarized in Table S1A–S1F, a precise delineation of the gene content has been performed in 15 cases, and dosage-sensitive candidate genes for the phenotype have been identified in only a few cases [19,20,22,113]. However, some general observations can be made (see Table 1). Of the 21 prenatal cases, there were at least 8 with ultrasound abnormalities (38%), of which 6 were terminated or resulted in spontaneous abortion or stillbirth. Of the 40 postnatal cases in which sufficient clinical information was available, MCA/MR was reported in 31 cases (77.5%). Speech delay was noted in 7 cases (17.5%), seizures in 2 cases (5%) and autism in 2 cases (5%). Multiple congenital abnormalities were present in 35 cases (87.5%) and were of variable severity. Dysmorphic signs were reported in 30 cases (75%), cardiac abnormalities in 7 cases (17.5%), a short stature in 6 cases (15%), and there was one case with abnormalities of the kidney (2.5%). We conclude that mental retardation or developmental delay of variable severity is present in about 80% of postnatal cases with multiple sSMCs. About 90% of postnatal cases have multiple congenital abnormalities or dysmorphic features.
Clinically Normal Phenotypes in Patients with Multiple sSMCs
In general, 70% of the carriers of a de novo, single sSMC are clinically normal [6]. It is unknown how many patients with multiple sSMCs have a normal phenotype, but this is expected to be lower than 70% because in cases with multiple sSMCs pathogenic genomic imbalances are more likely to occur compared to cases with single sSMC. Of the 40 postnatal cases with information on the phenotype (Table S1), 5 concern apparently normal individuals (12.5%, see Table S2), who were karyotyped because of a chromosome abnormality in a child, recurrent abortion or infertility. Remarkably, 8 of the 21 prenatal cases (38%) were reported to have no ultrasound abnormalities, and 6 of these 8 cases (29%) resulted in the birth of a normal, healthy baby (Table S2). In the 2 other prenatal cases there were up to 7 sSMCs. In one of these, when examined after birth, the male baby had unilateral hydronephrosis as the only clinical sign. There were no malformations and no dysmorphic signs except mild macrocephaly (which was considered a familial trait), and the boy showed normal development at 1 year of age [114]. The other case with up to 7 sSMCs concerned a boy who was reported to be of normal development at 2 years of age, with hypospadias and undescended testis as the only clinical abnormalities [115].
Although in most of the cases with a normal phenotype the gene content of the sSMCs has not been precisely identified, the sSMCs are relatively small. Therefore, we propose that the most likely explanation for the absence of clinical signs is that the pericentromeric regions contained within the sSMCs are located within the so-called “uncritical region” of the respective chromosome. In addition, somatic mosaicism with a low proportion of cells with the sSMCs is expected to contribute to a (near) normal phenotype [108]. This is exemplified by at least one of the cases in Table S2 (case 23 in reference [13]), with ~94% of amniocyte metaphases having a normal karyotype. Another example is provided by case mult 2-26, provided by M. Sagai, Israel (see http://ssmc-tl.com/sSMC.html), of a woman carrying a r(7)(::p11.2→q11.21::) and a min(13) (pter→q12.1:), who has mild mental retardation and is slightly dysmorphic, but who is completely functioning as an adult, “working in an office job, looking after a household, and a very conscientious mother” [14]. Both sSMCs probably contain genes located distally from the uncritical regions of chromosome 7 and 13, respectively, but the near-normal phenotype in this case may be explained by mosaicism, with the r(7) being present in only 11% of buccal mucosa cells, and 50% of cultured lymphocytes lacking one of the two sSMCs.
In conclusion, 12.5% of postnatal and 38% of prenatal cases with multiple sSMCs show a normal or mild clinical phenotype. These percentages are surprisingly high and have important consequences for genetic counseling, for example when a karyotype with multiple sSMCs is identified during prenatal diagnosis. In such situations it is imperative to both accurately delineate the genomic imbalances and to investigate a sufficiently high number of cells to determine mosaicism of the sSMCs, so that the outcome of the pregnancy can be predicted as accurately as possible.
Genetic Diagnostic Testing Summary
Most cases with multiple sSMCs are detected during the routine, array-based genome profiling of a fetus or individual who harbors a complex set of phenotypes that is not reminiscent of a known syndrome. In all cases in which parental karyotypes were determined, the multiple sSMCs arose de novo. This indicates that the recurrence risk for multiple sSMCs is very low (although the rare possibility of germ line mosaicism cannot be completely excluded). If the sSMCs contain pericentromeric euchromatin, a duplication signal as a “signature” of the sSMC will appear on the array data plot. The latter not only facilitates detection of sSMCs, but also allows to precisely determine the size of the sSMCs. If the sSMCs extend beyond the uncritical region of the corresponding chromosome (see http://ssmc-tl.com/sSMC.html), dosage-sensitive candidate genes for the phenotype can be identified. Given that somatic mosaicism of the multiple sSMCs occurs in more than 90% of the cases in which this was studied, the distribution of the different sSMCs in various tissues must be analysed as well by investigating a sufficiently high number of cells, either by karyotyping or FISH. For example, a sSMC that occurs in only a minority of cells is expected to contribute less to the clinical abnormalities seen in the patient. Together with the gene content, the degree of mosaicism of each sSMC is also important for deriving genotype-phenotype correlations. Finally, UPD of the structurally normal homologous chromosomes must be excluded.
An unexpected finding from this review is that ~12.5% of postnatal cases have no clinical abnormalities. Even more surprising is that ~38% of cases ascertained before birth did not show ultrasound abnormalities and that ~29% of these prenatal cases resulted in the birth of a normal, healthy baby. This shows that the termination of a pregnancy with a fetus carrying multiple sSMCs must be considered with the utmost care. In such cases accurate information on the gene content and mosaicism of the different sSMCs is critical for adequate clinical management of the pregnancy.
Author Contributions
All authors contributed to the data collection and analysis and the writing of this review.
Competing Interests
The authors have declared that no competing interests exist.
References
- Crolla JA, Youings SA, Ennis S, Jacobs PA. Supernumerary marker chromosomes in man: parental origin, mosaicism and maternal age revisited. Eur J Hum Genet. 2005;13:154-60. [CrossRef]
- Starke H, Nietzel A, Weise A, Heller A, Mrazek K, Belitz B, et al. Small supernumerary marker chromosomes (sSMCs): genotype-phenotype correlation and classification. Hum Genet. 2003;114:51-67. [CrossRef]
- Liehr T, Claussen U, Starke H. Small supernumerary marker chromosomes (sSMC) in humans. Cytogenet Genome Res. 2004;107:55-67. [CrossRef]
- Liehr T. Small supernumerary marker chromosomes (sSMC). A guide for human geneticists and clinicians. Berlin: Springer Verlag; 2012;p. 220. [CrossRef]
- Jafari-Ghahfarokhi H, Moradi-Chaleshtori M, Liehr T, Hashemzadeh-Chaleshtori M, Teimori H, Ghasemi-Dehkordi P. Small supernumerary marker chromosomes and their correlation with specific syndromes. Adv Biomed Res. 2015;4:140;DOI: 10.4103/2277-9175.161542. [CrossRef]
- Liehr T, Weisse A. Frequency of small supernumerary marker chromosomes in prenatal, newborn, developmentally retarded and infertility diagnostics. Int J Mol Med. 2007;19:719-31. [CrossRef]
- Yu S, Fiedler SD, Brawner SJ, Joyce JM, Zhou XG, Liu HY. Characterizing small supernumerary marker chromosomes with combination of multiple techniques. Cytogenet Genome Res. 2012;136:6-14. [CrossRef]
- Vetro A, Manolakos E, Petersen MB, Thomaidis L, Liehr T, Croci G, et al. Unexpected results in the constitution of small supernumerary marker chromosomes. Eur J Med Genet. 2012;55:185-90. [CrossRef]
- Liehr T, Cirkovic S, Lalic T, Guc-Scekic M, de Almeida C, Weimer J, et al. Complex small supernumerary marker chromosomes - an update. Mol Cytogenet. 2013;6:46; DOI: 10.1186/1755-8166-6-46. [CrossRef]
- Reddy KS, Aradhya S, Meck J, Tiller G, Abboy S, Bass H. A systematic analysis of small supernumerary marker chromosomes using array CGH exposes unexpected complexity. Genet Med. 2013;15:3-13. [CrossRef]
- Malvestiti F, De Toffol S, Grimi B, Cinetti S. Marcato L, Agrati C, et al: De novo small supernumerary marker chromosomes detected on 143,000 consecutive prenatal diagnoses: chromosomal distribution, frequencies, and characterization combining molecular cytogenetics approaches. Prenat Diagn. 2014;34:460-68. [CrossRef]
- Liehr T, Ewers E, Hamid AB, Kosyakova N, Voigt M, Weise A, Manvelyan M. Small supernumerary marker chromosomes and uniparental disomy have a story to tell. J Histochem Cytochem. 2011;59:842-848. [CrossRef]
- Baldwin EL, May LF, Justice AN, Martin CL, Ledbetter DH. Mechanisms and consequences of small supernumerary marker chromosomes: from Barbara McClintock to modern geneticcounseling issues. Am J Hum Genet. 2008;82:398-410. [CrossRef]
- Liehr T. Small supernumerary marker chromosomes [Internet]. Jena, Germany; 2016 [cited 2016, October 24th]. Available from: http://ssmc-tl.com/sSMC.html.
- Beverstock GC, Bezrookove V, Mollevanger P, van de Kamp JJP, Pearson P, Kouwenberg JM, Rosenberg C. Multiple supernumerary ring chromosomes of different origin in a patient: a clinical report and review of the literature. Am J Med Genet Part A. 2003;122A:168-73. [CrossRef]
- Daniel A, Malafiej P. A series of supernumerary small ring marker autosomes identified by FISH with chromosome probe arrays and literature review excluding chromosome 15. Am J Med Genet. 2003;117A:212-22. [CrossRef]
- Reddy KS, Wang S, Groh S, Gonatos J. SKY assessment of two karyotypes with 0-6 supernumerary marker/ring chromosomes and review of previously reported cases with two or more markers. Am J Med Genet. 2003;118A:156-71. [CrossRef]
- Golubovsky MD. Postzygotic diploidization of triploids as a source of unusual cases of mosaicism, chimerism and twinning. Hum Reprod. 2003;18:236-42. [CrossRef]
- Kogan JM, Miller E, Ware SM: High resolution SNP based microarray mapping of mosaic supernumerary marker chromosomes 13 and 17: delineating novel loci for apraxia. Am J Med Genet Part A. 2009;149A:887-93. [CrossRef]
- Hochstenbach R, van Gijn M, Krijtenburg PJ, Raemakers R, van ‘t Slot R, Renkens I, et al. Gain of FAM123B and ARHGEF9 in an obese man with intellectual disability, congenital heart defects and multiple supernumerary ring chromosomes. Mol Syndromol. 2013;3:274-83. [CrossRef]
- Schwanitz G, Hagh JK, Rad IA, Omrani MD, Gamerdinger U, Schubert R, et al. Patient with three euchromatic supernumerary marker chromosomes derived from chromosomes 1, 12, and 18: characterization and evaluation of the aberrations. Am J Med Genet Part A. 2014;164A:736-40. [CrossRef]
- Hochstenbach R, Nowakowska B, Volleth M, Ummels A, Kutkowska-Kaźmierczak A, Obersztyn E, et al. Multiple small supernumerary marker chromosomes resulting from maternal meiosis I or II errors. Mol Syndromol. 2016;6:210-21. [CrossRef]
- Viersbach R, Engels H, Gamerdinger U, Hansmann M. Delineation of supernumerary marker chromosomes in 38 patients. Am J Med Genet. 1998;76:351-58. [CrossRef]
- Bartsch O, Loitzsch A, Kozlowski P, Mazauric M-L, Hickmann G. Forty-two supernumerary marker chromosomes (SMCs) in 43 273 prenatal samples: chromosomal distribution, clinical findings, and UPD studies. Eur J Hum Genet. 2005;13:1192-204. [CrossRef]
- Plattner R, Heerema NA, Howard-Peebles PN, Miles JH, Soukup S, Palmer CG. Clinical findings in patients with marker chromosomes identified by fluorescence in situ hybridisation. Hum Genet. 1993;91:589-98. [CrossRef]
- Shanske AL, Dowling P, Schmidt R, White BJ, Russell B, Bogdanow A, Marion RW. Simultaneous occurrence of two supernumerary autosomal ring chromosomes r(1) and r(16) in twins. J Med Genet. 1999;36:625-8.
- Ballif BC, Rorem EA, Sundin K, Lincicum M, Gaskin S, Coppinger J, et al. Detection of low-level mosaicism by array CGH in routine diagnostic specimens. Am J Med Genet A. 2006;140:2757-67. [CrossRef]
- Cheung SW, Shaw CA, Scott DA, Patel A, Sahoo T, Bacino CA, et al. Microarray-based CGH detects chromosomal mosaicism not revealed by conventional cytogenetics. Am J Med Genet Part A. 2007;143A:1679-86. [CrossRef]
- Conlin LK, Thiel BD, Bonnemann CG, Medne L, Ernst LM, Zackai EH, et al. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum Mol Genet. 2010;19:1263-75. [CrossRef]
- Bruno DL, White SM, Ganesamoorthy , Burgess T, Butler K, Corrie S, et al. Pathogenic aberrations revealed exclusively by single nucleotide polymorphism (SNP) genotyping data in 5000 samples tested by molecular karyotyping. J Med Genet. 2011;48:831-9. [CrossRef]
- Sheth F, Andrieux J, Ewers E, Kosyakova N, Weise A, Sheth H, et al. Characterization of sSMC by FISH and molecular techniques. Eur J Med Genet. 2011;54:247-55. [CrossRef]
- Vermeesch JR, Duhamel H, Petit P, Falzetti D, Fryns J-P, Marynen P. Multiple small accessory marker chromosomes from different centromeric origin in a moderately mentally retarded male. Hum Genet. 1999;105:611-8. [CrossRef]
- Liehr T, Starke H, Senger G, Melotte C, Weise A, Vermeesch JR. Overrepresentation of supernumerary marker chromosomes (sSMC) from chromosome 6 origin in cases with multiple sSMC. Am J Med Genet. 2006;140A:46-51. [CrossRef]
- Angell RR. Chromosome abnormalities in human preimplantation embryos. Prog Clin Biol Res. 1989;294:181-7.
- McFadden DE, Langlois S. Parental and meiotic origin of triploidy in the embryonic and fetal periods. Clin Genet. 2000;58:192-200. [CrossRef]
- McFadden DE, Robinson WP. Phenotype of triploid embryos. J Med Genet 2006;43:609-12. [CrossRef]
- Grossmann M, Calafell JM, Brandy N, Vanrell JA, Rubio C, Pellicer A, et al. Origin of tripronucleate zygotes after intracytoplasmic sperm injection. Hum Reprod. 1997;12:2762-5. [CrossRef]
- Chen Z, Yan J, Feng HL. Aneuploid analysis of tripronuclear zygotes derived from in vitro fertilization and intracytoplasmic sperm injection in humans. Fertil Steril. 2005;83:1845-8. [CrossRef]
- Rosenbusch BE. Mechanisms giving rise to triploid zygotes during assisted reproduction. Fertil Steril. 2008;90:49-55. [CrossRef]
- Rosenbusch BE. A preliminary concept, deduced from cytogenetic analyses, for explaining different types of multipronuclear oocytes obtained after intracytoplasmic sperm injection. Fertil Steril. 2010;94:2479-81. [CrossRef]
- Rosenbusch BE, Schneider M. Hypotroploid tripronuclear oocytes with two polar bodies obtained after ICSI: Is irregular chromatid segregation involved? Hum Reprod. 2000;15:1876-7. [CrossRef]
- Rosenbusch BE, Schneider M: Separation of a pronucleus by premature cytokinesis: a mechanism for immediate diploidization of tripronuclear oocytes? Fertil Steril. 2009;92:394.e5-e8. DOI: 10.1016/j.fertnstert.2009.02.037. [CrossRef]
- Fragouli E, Alfarawati S, Spath K, Jaroudi S, Sarasa J, Enciso M, Wells D. The origin and impact of embryonic aneuploidy. Hum Genet. 2013;132:1001-13. [CrossRef]
- Coonen E, Derhaag JG, Dumoulin JCM, van Wissen LCP, Bras M, Janssen M, et al. Anaphase lagging mainly explains chromosomal mosaicism in human preimplantation embryos. Hum Reprod. 2004;19:316-24. [CrossRef]
- Daphnis DD, Delhanty JDA, Jerkovic S, Geyer J, Craft I, Harper JC. Detailed FISH analysis of day 5 human embryos reveals the mechanisms leading to mosaic aneuploidy. Hum Reprod. 2005;20:129-37. [CrossRef]
- McCoy RC, Demko Z, Ryan A, Banjevic M, Hill M, Sigurjonson S, et al. Common variants spanning PLK4 are associated with mitotic-origin aneuploidy in human embryos. Science. 2015;348:235-8. [CrossRef]
- Ioannou D, Fonseka KGL, Meershoek EJ, Thornhill AR, Abogrein A, Ellis M, Griffin DK. Twentyfour chromosome FISH in human IVF embryos reveals patterns of post-zygotic chromosome segregation and nuclear organization. Chromosome Res. 2012;20:447-60. [CrossRef]
- Capalbo A, Bono S, Spizzichino L, Biricik A, Baldi M, Colamaria S, et al. Sequential comprehensive chromosome analysis on polar bodies, blastomeres and throphoblast: insights into female meiotic errors and chromosomal segregation in the preimplantation window of embryo development. Hum Reprod.2013;28:509-18. [CrossRef]
- Voet, T, Vanneste E, Vermeesch J. The human cleavage stage embryo is a cradle of chromosomal rearrangements. Cytogenet Genome Res. 2011;133:160-8. [CrossRef]
- Li Q-Y, Tsukishiro S, Nakagawa C, Tanemura M, Sugiura-Ogasawara M, Suzumori K, Sonta S. Parental origin and cell stage of non-disjunction of double trisomy in a spontaneous abortion. Congenit Anom. 2005;45:21-5. [CrossRef]
- Fragouli E, Alfarawati S, Goodall N-n, Sánchez-Garcia JF, Colls P, Wells D. The cytogenetics of polar bodies: Insights into female meiosis and the diagnosis of aneuploidy. Mol Hum Reprod. 2011;17:286-95. [CrossRef]
- Handyside AH, Montag M, Magli MC, Repping S, Harper J, Schmutzler A, et al. Multiple meiotic errors caused by predivision of chromatids in women of advanced maternal age. Eur J Hum Genet. 2012;20:742-7. [CrossRef]
- Fragouli E, Wells D. Human embryonic aneuploidy. In: eLS. Chichester: John Wiley & Sons, Ltd; 2014. DOI: 10.1002/9780470015902.a0025706. [CrossRef]
- Ottolini CS, Newnham LJ, Capalbo A, Natesan SA, Joshi HA, Cimadomo D, et al. Genome-wide maps of recombination and chromosome segregation in human oocytes and embryos show selection for maternal recombination rates. Nat Genet. 2015;47:727-35. [CrossRef]
- Verpoest W, Fauser BC, Papanikolaou E, Staessen C, Van Landuyt L, Donoso P, et al. Chromosomal aneuploidy in embryos conceived with unstimulated cycle IVF. Human Reprod. 2008;23:2369–71. [CrossRef]
- Labarta E, Bosch E, Alama´ P, Rubio C, Rodrigo L, Pellicer A. Moderate ovarian stimulation does not increase the incidence of human embryo chromosomal abnormalities in in vitro fertilization cycles. J Clin Endocrinol Metab. 2012;97:E1987–E1994. DOI: 10.1210/jc.2012-1738. [CrossRef]
- Hou Y, Fan W, Yan L, Li R, Lian Y, Huang J, et al. Genome analysis of single human oocytes. Cell. 2013;155:1492-506. [CrossRef]
- Reddy KS. Double trisomy in spontaneous abortions. Hum Genet. 1997;101:339-45. [CrossRef]
- Reddy KS. Triple aneuploidy in spontaneous abortions. Clin Genet. 1999;56:103-4. [CrossRef]
- Micale M, Insko J. Ebrahim SA, Adeyinka A, Runke C, Van Dyke DL. Double trisomy revisited —A multicenter experience. Prenat Diagn. 2010;30:173-6. [CrossRef]
- Subramaniyam S, Pulijaal VR, Mathew S. Double and multiple chromosomal aneuploidies in spontaneous abortions: A single institutional experience. J Hum Reprod Sci. 2014;7:262-8. [CrossRef]
- Miura K, Yoshiura K, Miura S, Kondoh T, Harada N, Yamasaki K, et al. Clinical outcome of infants with confined placental mosaicism and intrauterine growth restriction of unknown cause. Am J Med Genet Part A. 2006;140A:1827-33. [CrossRef]
- Diego-Alvarez D, Ramos-Corrales C, Garcia-Hoyos M, Bustamante-Aragones A, Cantalapiedra D, Diaz-Recasens J, et al. Double trisomy in spontaneous miscarriages: cytogenetic and molecular approach. Hum Reprod. 2006;21:958-66. [CrossRef]
- Vogt E, Kirsch-Volders M, Parry J, Eichenlaub-Ritter U. Spindle formation, chromosome segregation and the spindle assembly checkpoint in mammalian oocytes and susceptibility to meiotic error. Mutat Res. 2008; 651:14-29. [CrossRef]
- Nagaoka K, Hassold TJ, Hunt PA. Human aneuploidy: mechanisms and new insights into an age-old problem. Nat Rev Genet. 2012;13:493-504. [CrossRef]
- MacLennan M, Crichton JH, Playfoot CJ, Adams IR. Oocyte development, meiosis and aneuploidy. Semin Cell Dev Biol. 2015;45:68-76. [CrossRef]
- Herbert M, Kalleas D, Cooney D, Lamb M, Lister L. Meiosis and maternal ageing: insights from aneuploidy oocytes and trisomy births. Cold Spring Harb Perspect Biol. 2015;7:a017970. DOI: 10.1101/cshperspect.a017970. [CrossRef]
- Angell RR. Predivision in human oocytes at meiosis I: A mechanism for trisomy formation in man. Hum Genet. 1991;86:383-7. [CrossRef]
- Lister LM, Kouznetsova A, Hyslop LA, Kalleas D, Pace SL, Barel JC, et al. Age-related meiotic segregation errors in mammalian oocytes are preceded by depletion of cohesion and Sgo2. Curr Biol. 2010;20:1511-21. [CrossRef]
- Yun Y, Holt JE, Lane SIR, McLaughlin EA, Merriman JA, Jones KT. Reduced ability to recover from spindle disruption and loss of kinetochore spindle assembly checkpoint proteins in oocytes from aged mice. Cell Cycle. 2014;13:1939-47. [CrossRef]
- Jones KT, Lane SIR. Molecular causes of aneuploidy in mammalian eggs. Development. 2013;140:3719-30. [CrossRef]
- Broman KW, Murray JC, Sheffield VC, White RL, Weber JL. Comprehensive human genetic map: individual and sex-specific variation in recombination. Am J Hum Genet 1998;63:861-9. [CrossRef]
- Cheung VG, Burdick JT, Hirschmann D, Morley M. Polymorphic variation in human meiotic recombination. Am J Hum Genet. 2007;80:526-30. [CrossRef]
- Coop G, Wen X, Ober C. Pritchard JK, Przeworski M. High-resolution mapping of crossovers reveals extensive variation in fine-scale recombination patterns among humans. Science. 2008;319:1395-8. [CrossRef]
- Lynn A, Ashley T, Hassold T. Variation in human meiotic recombination. Annu Rev Genomics Hum Genet. 2004;5:317-49. [CrossRef]
- Delhanty JD. Inherited aneuploidy: germline mosaicism. Cytogenet Genome Res. 2011;133:136-40. [CrossRef]
- Ghevaria H, SenGupta S, Sarna U, Sargeant S, Serhal P, Delhanty J. The contribution of germinal mosaicism to human aneuploidy. Cytogenet Genome Res. 2014;144:264-74. [CrossRef]
- Kovaleva NV, Tahmasebi-Hesari M. Gonadal mosaicism. Down Syndrome News. 2007;14:23.
- Rowsey R, Kashevarova A, Murdoch B, Dickenson C, Woodruff T, Cheng E, et al. Germline mosaicism does not explain the maternal age effect on trisomy. Am J Med Genet Part A. 2013;161A:2495-503. [CrossRef]
- Templado C, Uroz L, Estop A. New insights on the origin and relevance of aneuploidy in human spermatozoa. Mol Hum Reprod. 2013;19:634-43. [CrossRef]
- Norris-Kirby A, Hagenkord JM, Kshirsagar MP, Ronnett BM, Murphy KM. Abnormal villous morphology associated with triple trisomy of paternal origin. J Mol Diagn. 2010;12:525-9. [CrossRef]
- Rubes J, Vozdova M, Oracova E, Perrault SD. Individual variation in the frequency of sperm aneuploidy in humans. Cytogenet Genome Res. 2005;111:229-36. [CrossRef]
- Tempest HG, Ko E, Rademaker A, Chan P, Robaire B, Martin RH. Intra-individual and interindividual variations in sperm aneuploidy frequencies in normal men. Fertil Steril. 2009;91:185-92. [CrossRef]
- Tomascik-Cheeseman LM, Lowe XR, Eskenazi B, Kidd S, Nath J, Moore D, Wyrobek AJ. A father of four consecutive trisomic pregnancies with elevated frequencies of associated aneuploid sperm. Am J Med Genet Part A. 2006;140A:1840-5. [CrossRef]
- Thomas NS, Hassold TJ. Aberrant recombination and the origin of Klinefelter syndrome. Hum Reprod Update. 2003;9:309-17. [CrossRef]
- Ballif BC, Hornor SA, Sulpizio SG, Lloyd RM, Minier SL, Rorem EA, et al. Development of a high-density pericentromeric region BAC clone set for the detection and characterization of small supernumerary marker chromosomes by array CGH. Genet Med. 2007;9:150-62. [CrossRef]
- Castronovo C, Valtorta E, Crippa M, Tedoldi S, Romitti L, Amione MC, et al. Design and validation of a pericentromeric BAC clone set aimed at improving diagnosis and phenotype prediction of supernumerary marker chromosomes. Mol Cytogenet. 2013;6:45. DOI: 10.1186/1755-8166-6-45. [CrossRef]
- Hamid AB, Fan X, Kosyakova N, Radhakrishnan G, Liehr T, Karamysheva T. New BAC probe set to narrow down chromosomal breakpoints in small and large derivative chromosomes, especially suited for mosaic conditions. Methods Mol Biol. 2015;1227:279-87. [CrossRef]
- Martínez-A C, van Wely KHM. Are aneuploidy and chromosome breakage caused by a CINgle mechanism? Cell Cycle. 2010;9:2275-80. [CrossRef]
- Guerrero AA, Gamero MC, Trachana V, Fütterer A, Pacios-Bras C, Díaz-Concha NP, et al. Centromere-localized breaks indicate the generation of DNA damage by the mitotic spindle. Proc Natl Acad Sci USA. 2010;107:4159-64. [CrossRef]
- Guerrero AA, Martínez-A, van Wely KHM. Merotelic attachments and non-homologous end joining are the basis of chromosomal instability. Cell Div. 2010;5:13, DOI: 10.1186/17471028-5-13. [CrossRef]
- Nambiar M, Smith GR. Repression of harmful meiotic recombination in centromeric regions. Semin Cell Dev Biol. 2016;54:188-97. [CrossRef]
- May KM, Jacobs PA, Lee M, Ratcliffe S, Robinson A, Nielsen J, Hassold TJ. The parental origin of the extra X-chromosome in 47,XXX females. Am J Hum Genet. 1990;46:754-61.
- Thomas NS, Ennis S, Sharp AJ, Durkie M, Hassold TJ, Collins AR, Jacobs PA. Maternal sexchromosome nondisjunction: evidence for X-specific risk factors. Hum Molec Genet. 2001;10:243-50. [CrossRef]
- Oliver TR, Feingold E, Yu K, Cheung C, Tinker S, Yadav-Shah M et al. New insights into human nondisjunction of chromosome 21 in oocytes. PLoS Genet. 2008;4:e1000033. DOI: 10.1371/journal.pgen.1000033. [CrossRef]
- Wevrick R, Earnshaw WC, Howard-Peebles PN, Willard HF. Partial deletion of alpha satellite DNA associated with reduced amounts of the centromere protein CENP-B in a mitotically stable human chromosome rearrangement. Molec Cell Biol. 1990;10:6374-80. [CrossRef]
- Yang JW, Pendon C, Yang J, Haywood N, Chand A, Brown WRA. Human mini-chromosomes with minimal centromeres. Hum Molec Genet. 2000;9:1891-902. [CrossRef]
- Aldrup-MacDonald ME, Sullivan BE. The past, present, and future of human centromere genomics. Genes. 2014;5:35-50. [CrossRef]
- Liehr T. Benign and pathological chromosomal imbalances. Microscopic and submicroscopic copy number variations (CNVs) in genetics and counseling. Amsterdam: Academic Press;2014.p. 187.
- Ma J-Y, Yang Y-C O, Wang Z-W, Wang Z-B, Jiang Z-Z, Luo SM, et al. The effects of DNA doublestrand breaks on mouse oocyte meiotic maturation. Cell Cycle. 2013;12:1233-41. [CrossRef]
- Lin F, Ma XS, Wang ZB, Wang ZW, Luo YB, Huang L, et al. Different fates of oocytes with DNA double-strand breaks in vitro and in vivo. Cell Cycle. 2014;13:2674-80. [CrossRef]
- Poot M, Haaf T. Mechanisms of origin, phenotypic effects and diagnostic implications of complex chromosome rearrangements. Mol Syndromol. 2015;6:109-33. [CrossRef]
- Tsuchiya KD, Opheim KE, Hannibal MC, Hing AV, Glass IA, Raff ML, et al. Unexpected structural complexity of supernumerary marker chromosomes characterized by microarray comparative genomic hybridization. Mol Cytogenet. 2008;1:7. DOI: 10.1186/1755-8166-1-7. [CrossRef]
- Davidsson J, Jahnke K, Forsgren M, Collin A, Soller M. dup(19)(q12q13.2): array-based genotype-phenotype correlation of a new possibly obesity-related syndrome. Obesity. 2010;18:580-7. [CrossRef]
- Hu J, Madan-Khetarpal S, Serrano Russi AH, Kochmar S, Deward SJ, Sathanoori M, Surti U. Three supernumerary marker chromosomes in a patient with developmental delay, mental retardation, and dysmorphic features. Genet Res Int. 2011;2011:185271. DOI: 10.4061/2011/185271. [CrossRef]
- Hussein SS, Kreskowski K, Ziegler M, Klein E, Hamid AB, Kosyakova N, et al: Mitotic stability of small supernumerary marker chromosomes depends on their shape and telomeres—A long term in vitro study. Gene. 2014;552:246-8. [CrossRef]
- Spittel H, Kubek F, Kreskowski K, Ziegler M, Klein E, Hamid AB, et al. Mitotic stability of small supernumerary marker chromosomes: a study based on 93 immortalized cell lines. Cytogenet Genome Res. 2014;142:151-60. [CrossRef]
- Liehr T, Klein E, Mrasek K, Kosyakova N, Guilherme RS, Aust N, et al. Clinical impact of somatic mosaicism in cases with small supernumerary marker chromosomes. Cytogenet Genome Res. 2013;139:158-63. [CrossRef]
- Hochstenbach R, van Binsbergen E, Engelen J, Nieuwint A, Polstra A, Poddighe P, et al. Array analysis and karyotyping: workflow consequences based on a retrospective study of 36,325 patients with idiopathic developmental delay in the Netherlands. Eur J Med Genet. 2009;52:161-9. [CrossRef]
- Poot M, Hochstenbach R. A three-step workflow procedure for the interpretation of arraybased comparative genome hybridization results in patients with idiopathic mental retardation and congenital anomalies. Genet Med. 2010;12:478-85. [CrossRef]
- Papadopoulos T, Soykan T. The role of collybistin in gephyrin clustering at inhibitory synapses: facts and open questions. Front Cell Neurosci. 2011;5:11. DOI: 10.3389/fncel.2011.00011. [CrossRef]
- Soykan T, Schneeberger D, Tria G, Buechner C, Bader N, Svergun D, et al. A conformational switch in collybistin determines the differentiation of inhibitory postsynapses. EMBO J. 2014;33:2113-33. [CrossRef]
- Guediche N, Tosca L, Kara Terki A, Bas C, Lecerf L, Young J, et al. Array comparative genomic hybridization analysis of small supernumerary marker chromosomes in human infertility. Reprod Biomed Online. 2012;24:72-82. [CrossRef]
- Ulmer R, Pfeiffer RA, Wiest E, Goelz R, Trautmann U. Multiple (up to seven) different accessory marker chromosomes: prenatal diagnosis and follow-up. Ann Génét. 1997;40:109-14.
- Chen M, Chang SP, Yin PL, Sapeta M, Barringer S, Kuo SJ, et al. Prenatal identification of small supernumerary marker chromosomes by FISH in an infant born with mild congenital anomalies. Prenat Diagn. 2006;26:383-7. [CrossRef]
- Callen DF, Eyre HJ, Ringenbergs ML, Freemantle CJ, Woodroffe P, Haan EA. Chromosomal origin of small ring marker chromosomes in man: characterization by molecular genetics. Am J Hum Genet. 1991;48:769-82.
- Wiktor A, Van Dyke DL, Weiss L. Characterization of a de novo 48,XX,+r(X),r(17) by in situ hybridization in a patient with neurofibromatosis (NF1). Am J Med Genet. 1993;45:22-4. [CrossRef]
- Aalfs CM, Jacobs ME, Nieste-Otter MA, Hennekam RCM, Hoovers JMN. Two supernumerary marker chromosomes, derived from chromosome 6 and 9, in a boy with mild developmental delay. Clin Genet. 1996;49:42-5. [CrossRef]
- Haddad BR, Schröck E, Meck J, Cowan J, Young H, Ferguson-Smith MA, et al. Identification of de novo chromosomal markers and derivatives by spectral karyotyping. Hum Genet. 1998;103:619-25. [CrossRef]
- Nandi KN, McDonald MT, Rogers KK, Rao KW. Mosaicism for two de novo supernumerary markers derived from chromosomes 18 and 13. Am J Hum Genet. 2001;69(Suppl):Abstract 793.
- Maurer B, Haaf T, Stout K, Reismann N, Steinlein C, Schmid M. Two supernumerary marker chromosomes, originating from chromosomes 6 and 11, in a child with developmental delay and craniofacial dysmorphism. Cytogenet Cell Genet. 2001;98:182-7. [CrossRef]
- Levy B, Jalal SM, Dunn TM, Warburton PE, Tonk VS, Hirschhorn K, et al. Unique case of mosaicism involving two morphologically similar marker chromosomes of different centric origin in a patient with developmental delay. Am J Med Genet. 2002;108:198-204. [CrossRef]
- Hall S, Boda Y, Cohen M, Junio J, Polihronis A, Tyrrell V, Wright D. FISH-mapping of r(9) and r(18) chromosomes in a mosaic newborn female. Chrom Res. 2005;13(Suppl. 1):61 (Abstract 1.108-P).
- Sanz R, Sousa A, Gonzalez S: Identification of the chromosomal origin of smal supernumerary marker chromosomes and its phenotypic effects. Chrom Res. 2005; 13(Suppl. 1):69 (Abstract 1.126-P).
- Weimer J, Metzke-Heidemann S, Plendl H, Caliebe A, Grunewald R, Ounap K, et al. Characterization of two supernumerary marker chromosomes in a patient with signs of Klinefelter syndrome, mild facial anomalies, and severe speech delay. Am J Med Genet. 2006;140A:488-95. [CrossRef]
- Huang B, Solomon S, Thangavelu M, Peters K, Bhatt S. Supernumerary marker chromosomes detected in 100 000 prenatal diagnoses: molecular cytogenetic studies and clinical significance. Prenat Diagn. 2006;26:142-50. [CrossRef]
- Pietrzak J, Mrasek K, Obersztyn E, Stankiewicz P, Kosyakova N, Weise A, et al. Molecular cytogenetic characterization of eight small supernumerary marker chromomomes originating from chromosomes 2, 4, 8, 18, and 21 in three patients. J Appl Genet. 2007;48:167-75. [CrossRef]
- Tönnies H, Pietrzak J, Bocian E, MacDermont K, Kuechler A, Belitz B, et al. New immortalized cell lines of patients with small supernumerary marker chromosome: towards the establishment of a cell bank. J Histochem Cytochem. 2007;55:651-60. [CrossRef]
- Neill NJ, Torchia BS, Bejjani BA, Shaffer LG, Ballif BC. Comparative analysis of copy number detection by whole-genome BAC and oligonucleotide array CGH. Mol Cytogenet. 2010;3:11. DOI: 10.1186/1755-8166-3-11. [CrossRef]
- Zolotukhina TV, Shilova NV, Judina EV, Minzhenkova ME, Kozlova YO. Small supernumerary marker chromosomes detected prenatally. Eur J Hum Genet. 2013;21(Suppl. 2):592.
- Mackie-Ogilvie C, Waddle K, Mandeville J, Seller MJ, Docherty Z. Rapid identification of multiple supernumerary ring chromosomes with a new FISH technique. J Med Genet. 1997;34:912-6. [CrossRef]
- Toksoy G, Türköver B, Sayar C, Söylemez MA, Yardimci T et al. Unusual prenatal case with multiple marker chromosomes. Chrom Res. 2007;15(Suppl. 1):127 (Abstract 1.241-P).
- Fernández-Toral J, Rodríguez L, Plasencia A, Martínez-Frías ML, Ewers E, Hamid AB, et al. Four small supernumerary marker chromosomes derived from chromosomes 6, 8, 11 and 12 in a patient with minimal clinical abnormalities: a case report. J Med Case Rep. 2010;4:239. DOI: 10.1186/1752-1947-4-239. [CrossRef]
- Joziasse IC, van der Smagt JJ, Poot M, Hochstenbach R, Nelen MR, van Gijn M, et al. A duplication including GATA4 does not co-segregate with congenital heart defects. Am J Med Genet Part A. 2009;149A:1062-6. [CrossRef]
- Anguiano A, Wang BT, Wang SR, Boyar FZ, Mahon LW, El Naggar MM, et al. Spectral Karyotyping for identification of constitutional chromosomal abnormalities at a national reference laboratory. Mol Cytogenet. 2012;5:3. DOI: 10.1186/1755-8166-5-3. [CrossRef]
- Le Du N, Chambon P, Drouin-Garaud V, Jloy-Helas G, Mabboux P et al. Up to 6 supernumerary marker chromosomes in a child with mild developmental delay- Complex genetic counseling. Chrom Res. 2009;17(Suppl. 1):181 (Abstract: 11.10-P).
- Fei X, Qi M, Zhao Y, Li-Ling J. Identification and characterization of a complex pure mosaic of small supernumerary marker chromosomes involving 11p11.12 → q12.1 and 19p12 → q12 regions in a child featuring multiple congenital anomalies. Am J Med Genet A. 2011;155A:3116-21. [CrossRef]
- Penton AL, Burnside RD, Gadi I, Jaswaney V, Schwartz S, Phillips K, et al. Cytogenetic characterization of 2 patients with supernumerary marker chromosomes (sSMCs) derived from complex genetic rearrangements. Proceedings of the American Society of Human Genetics 64th Annual Meeting, 18-22 October 2014; San Diego, CA, USA. p. 807; Abstract 3160T (2014).
- Daniel A, Malafiej P, Preece K, Chia N, Nelson J, Smith M. Identification of a marker chromosomes in thirteen patients using FISH probing. Am J Med Genet. 1994;53:8-18. [CrossRef]
- Hook EB, Cross PK. Extra structurally abnormal chromosomes (ESAC) detected at amniocentesis: frequency in approximately 75,000 prenatal cytogenetic diagnoses and associations with maternal and paternal age. Am J Hum Genet. 1987;40:83-101.
- Vundinti BR, Korgaonkar S, Ghosh K. De novo origin of multiple small supernumerary marker chromosomes (sSMCs) in a child with intellectual disability and dysmorphic features. Gene. 2012;498:128-130. [CrossRef]
- Mascarello JT, Jones MC, Chambers SR. A patient with extreme variation in number and size of small marker chromosomes. Hum Genet. 1987;75:191-4. [CrossRef]
- Tozzi C, Calvieri F, Alesi L, Neri G. Multiple “marker” chromosomes: a novel cytogenetic finding in a patient with mental retardation and congenital anomalies. Am J Med Genet. 1988;29:353-9. [CrossRef]
- Zaragoza MV, Jacobs PA, James RS, Rogan P, Sherman S, Hassold T. Nondisjunction of human acrocentric chromosomes: studies of 432 trisomic fetuses and liveborns. Hum Genet. 1994;94:411-7. [CrossRef]
- Park VM, Bravo RR, Shulman LP. Double non-disjunction in maternal meiosis II giving rise to a fetus with 48,XXX,+21. J Med Genet. 1995;32:650-3. [CrossRef]
- Chen C-P, Chern S-R, Weh L-F, Chen W-L, Chen L-F, Wang W. Prenatal diagnosis and genetic analysis of double trisomy 48,XXX,+18. Prenat Diagn. 2000;20:750-3. [CrossRef]
- Robinson WP, McFadden DE, Stephenson MD. The origin of abnormalities in recurrent aneuploidy/polyploidy. Am J Hum Genet. 2001;69:1245-54. [CrossRef]
- Biselli M, Machado FB, Zampieri BL, Alves da Silva AF, Goloni-Bertollo EM, Haddad R, et al. Double aneuploidy (48,XXY,+21) of maternal origin in a child born to a 13-year-old mother: evaluation of the maternal folate metabolism. Genet Couns. 2009;20:225-34.
- Guzel A, Demirhan O, Pazarbasi A, Ozgunen FT, Kocaturk-Sel S, Tastemir D. Detection of parental origin and cell stage errors of a double nondisjunction in a fetus by QF-PCR. Genet Test Mol Biomarkers. 2009;13:73-7. [CrossRef]
- Chen C-P, Chern S-R, Chen C-Y, Wu P-C, Chen L-F, Pan C-W, Wang W. Double aneuploidy with Edwards-Klinefelter syndromes (48,XXY,+18) of maternal origin: prenatal diagnosis and molecular cytogenetic characterization in a fetus with athrogryposis of the left wrist and aplasia of the left thumb. Taiwan J Obstet Gynacol. 2011;50:479-84. [CrossRef]
- Sheth HJ, Munoz A, Sergi C, Pani J, Blouin JL, Sheth JJ, Sheth FJ. Triple-X syndrome in a trisomic Down syndrome child: both aneuploidies originated from the mother. Int J Hum Genet. 2011;11:51-3. [CrossRef]
- Kajii T, Ferrier A, Nikawa N, Takahara H, Ohama K, Avirachan S. Anatomic and chromosomal abnormalities in 639 spontaneous abortuses. Hum Genet. 1980;55:87-98. [CrossRef]
- Hassold T, Chiu D, Yamane J. Parental origin of autosomal trisomies. Ann Hum Genet. 1984;48:129-44. [CrossRef]
- Chavarro R. Dougherty S, Fazikas L. William J, David-Nelson M. Triple trisomy associated with hydatidiform placenta. Appl Cytogenet. 1990;16:60.
- Johnson MP, Drugan A, Koppitch FC, Uhlmann WR, Evans MI. Postmortem chorionic villus sampling is a better method for cytogenetic evaluation of early fetal loss than culture of abortion material. Am J Obstet Gynecol. 1990;163:1505-10. [CrossRef]
- Petrella R, Hirschhorn K, German J. Triple autosomal trisomy in a pregnancy at risk for Bloom’s syndrome. Am J Med Genet. 1991;40:316-8. [CrossRef]
- Pettenati MJ, Rao N. Triple trisomy in a 17-week old fetus. Hum Genet. 1991;87:239-40. [CrossRef]
- Soukup SW. Triple trisomy in a spontaneous abortion. Hum Genet. 1992;88:363. [CrossRef]
- Warburton D. De novo balanced chromosome rearrangements and extra marker chromosomes identified at prenatal diagnosis: clinical significance and distribution of breakpoints. Am J Hum Genet. 1991;49:995-1013.
- Jacobs PA, Browne C, Gregson N, Joyce C, White H. Estimates of the frequency of chromosome abnormalities detectable in unselected newborns using moderate levels of banding. J Med Genet. 1992;29:103-8. [CrossRef]
- Blennow E, Bui TH, Kristoffersson U, Vujic M, Annerén G, Holmberg E, Nordenskjöld M. Swedish survey on extra structurally abnormal chromosomes in 39 105 consecutive prenatal diagnoses: prevalence and characterization by fluorescence in situ hybridization. Prenat Diagn. 1994;14:1019-28. [CrossRef]
- Forabosco A, Pescesepe A, Santucci S. Incidence of non-age-dependent chromosomal abnormalities: a population-based study on 88965 amniocenteses. Eur J Hum Genet. 2009;17:897-903. [CrossRef]
- Hook EB, Hamerton JL. The frequency of chromosome abnormalities detected in consecutive newborn studies - differences between studies - Results by sex and by severity of phenotypic involvement. In: Hook EB, Porter IH, editors. Population Cytogenetics. Studies in Humans. NewYork: Academic Press; 1977.p. 63-79.
- Nielsen J, Wohlert M. Chromosome abnormalities found among 34,910 newborn children: results from a 13-year incidence study in Arhus, Denmark. Hum Genet. 1991;87:81-3. [CrossRef]
- Maeda T, Ohno M, Matsumoto A, Yoshihara K, Yabe N. A cytogenetic survey of 14,835 consecutive liveborns. Jap J Hum Genet. 1991;36:117-29. [CrossRef]
- Ravel C. Berthaut I, Bresson JL, Siffroi JP and the Genetics Commission of the French Federation of CECOS. Prevalence of chromosomal abnormalities in phenotypically normal and fertile adult males: large-scale survey of over 10 000 sperm donor karyotypes. Hum Reprod. 2006;21:1484-89. [CrossRef]

