

The Role of Epigenetics in Developmental Programming and the Developmental Origins of Health and Disease
Kazuki Mochizuki 1,* ![]() , Chihiro Imai 2
, Chihiro Imai 2![]() , Noriko Sato 2
, Noriko Sato 2![]() , Takeo Kubota 3
, Takeo Kubota 3![]()
- Department of Local Produce and Food Sciences, Faculty of Life and Environmental Sciences, University of Yamanashi, Yamanashi, Japan
- Department of Molecular Epidemiology, Medical Research Institute, Tokyo Medical and Dental University, Tokyo, Japan
- Department of Child Studies, Faculty of Child Studies, Seitoku University, Chiba, Japan
* Correspondence: Kazuki Mochizuki ![]() . Tel: + 81-55-220-8829; Fax: +81-55-220-8829.
. Tel: + 81-55-220-8829; Fax: +81-55-220-8829.
Received: June 20, 2017 | Accepted: August 31, 2017 | Published: October 20, 2017
OBM Genetics 2017, Volume 1, Issue 4 doi:10.21926/obm.genet.1704008
Academic Editors: Stéphane Viville, Aafke van Montfoort and Marcel Mannens
Recommended citation: Mochizuki K, Imai C, Sato N, Kubota T. The Role of Epigenetics in Developmental Programming and the Developmental Origins of Health and Disease. OBM Genetics 2017;1(4):008; doi:10.21926/obm.genet.1704008.
© 2017 by the authors. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
Abstract
A number of epidemiological studies have suggested that environmental stresses, such as malnutrition during the fetal period, can induce development of metabolic disorders, such as obesity, type 2 diabetes, and hypertension, and and even psychiatric disorders in later life. This theory model is known as the Developmental Origins of Health and Disease (DOHaD) theory, in which postulates that “epigenetic memories”, involving DNA methylation, histone modifications and microRNA expression, are induced by environmental stresses during development. For example, the binding of transcription factors to cis-elements within the promoters and enhancer regions of genes induces demethylation leading to and subsequent histone acetylation and transcriptional activation. Several lines of evidence suggest that binding of bromodomain-containing protein 4 and positive transcription elongation factor B to acetylated histones within “gene body” regions initiates transcriptional elongation, and while histone H3K36 methylation and DNA methylation within “gene body” these regions terminates this transcriptional elongation. This new epigenetic model of the gene body regulation can be applied to the regulation of metabolic genes, which respond to carbohydrate signals and are associated with energy balance. Recent studies indicate that these new gene body epigenetic mechanisms, as well as the classical promoter/enhancer mechanism, are responsible for regulation of epigenetic changes found in offspring that have been exposed to malnutrition during the fetal period. In this context, further understanding of epigenetic gene regulation during the fetal period based on the “gene body epigenetic model” during the fetal period should provide new preventive and therapeutic strategies for adult diseases encompassed by DOHaD theory.
Keywords
Epigenetics; histone acetylation; BRD4; gene body; DOHaD
Introduction
Lifestyle choices, including dietary habits, can lead to the development of metabolic diseases, including obesity and type 2 diabetes, hypertension, lipid abnormalities, metabolic syndrome, and associated complications. These lifestyle factors are also relevant in the development of psychiatric disorders. Recent studies have demonstrated that these metabolic diseases develop via accumulation of the consequences of lifestyle choices throughout life; not only during adulthood, but also during development, including the fetal period and childhood. Newborns with low birth weight (under 2,500 g) have a higher risk of death resulting from cardiovascular diseases [1], type 2 diabetes [2], and metabolic syndrome [3]. In addition, a cohort study conducted in the Netherlands revealed a higher incidence of obesity among subjects born to women who had been malnourished during a period of famine in the Second World War than among their siblings who were not born during a time of severe food shortage [4]. A cohort study conducted in China also showed a higher incidence of integration disorder syndrome among subjects born to women who had experienced malnutrition during famine than among the generations born before and after this period [5]. In addition, in Japan, the number of newborns with low birth weight has increased rapidly in recent decades, while the average birth weight has decreased [6]. Therefore, it can be speculated that the children born during recent decades in Japan have a higher risk of developing metabolic diseases later in life. One cause of this may be the adherence to extreme diets before and during pregnancy by Japanese women in recent years.
The Developmental Origins of Health and Disease (DOHaD) theory [7] postulates that a predisposition to metabolic diseases occurs through developmental programming that is regulated by exposure to environmental factors. Development of these metabolic diseases is considered to be caused by the long-term exposure of cells and tissues to environmental stimuli, such as hyper- or hyponutrition, during development as well as in adulthood. A candidate mechanism for the development of metabolic diseases is epigenetic memory of chromatin. The DOHaD theory is underpinned by epigenetics, rather than by changes in DNA sequence (genetics), because according to the DOHaD theory, diseases develop from stored memories of environmental stimulation during development. Here, we review the relationship between epigenetics and the developmental programming/DOHaD theory.
General Epigenetic Regulation
In general, transcriptional regulation is mediated via transcription factors bound to DNA sequences located upstream of the transcription initiation site, particularly at promoter and enhancer regions. Transcription is initiated by the binding of transcription factors to specific sites, known as cis-regulatory elements, located in promoter and enhancer regions. This binding recruits transcription initiation complexes containing the RNA polymerase II–TATA box binding protein to the TATA box, which subsequently activates the transcription initiation reaction (Figure 1A).
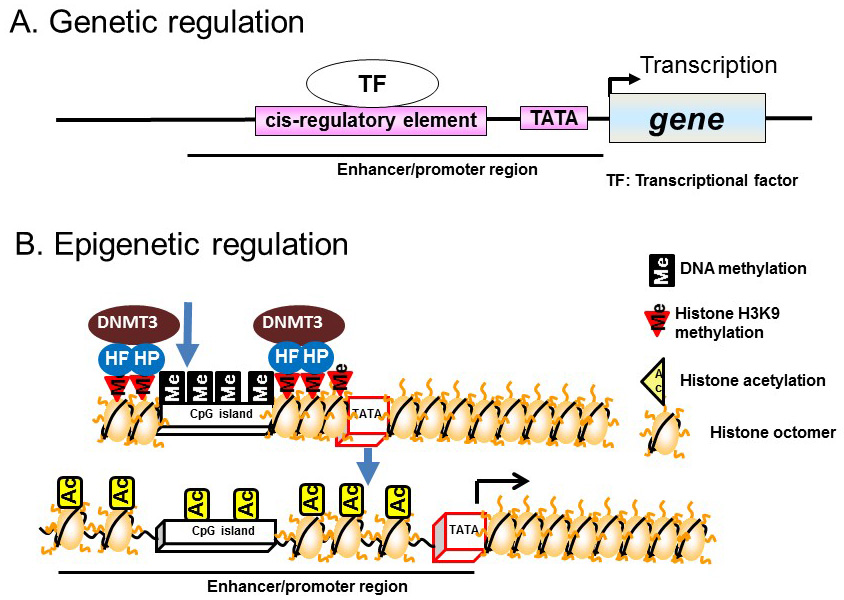
Figure 1 Genetic and epigenetic regulation. A) Genetic regulation. B) Epigenetic regulation.
Recent studies have demonstrated epigenetic regulation of transcription by DNA methylation of CpG dinucleotide-rich regions (CpG islands) in the promoter regions of genes. DNA methylation involves the addition of a methyl group to cytosine residues, particularly those within CpG islands. CpG islands hypermethylation leads to chromatin condensation by associating with heterochromatin binding proteins and DNA methyltransferases (DNMTs); this induces direct repression of transcription. Demethylation of CpG islands leads to the induction of transcription by altering histone modifications, such as acetylation, in enhancer/promoter regions (Figure 1B).
In contrast to the chemical modifications of DNA, those associated with chromosome histone proteins are rich in variety and include acetylation, methylation, phosphorylation, and ubiquitination (Figure 2). Histones form nucleosomes, which consist of a segment of DNA wound around a histone octamer containing two molecules each of H2A, H2B, H3, and H4. Methylation of histone H3 at lysine 4 (K4) generally confers transcriptional activation, while methylation of histone H3K9/K27/K36 and H4K20 is associated with transcriptional repression. Each histone methylation site can be mono-, di-, or trimethylated although, in general, transcriptional activation or repression is strongly associated with changes from mono- to trimethylation. Histone acetylation relaxes the chromatin structure by changing the histone charge. In addition, histone acetylation activates transcription by recruiting epigenetic “reader” or “writer” proteins. Reader proteins bind to sites of epigenetic modification and recruit a variety of complexes to chromatin, while writer proteins, such as histone acetyl-transferases (HATs), histone methyl-transferases, and DNMTs, induce epigenetic modifications of chromatin. Histone methylation does not alter the charge of histones, but induces changes in transcriptional activation/repression by recruiting methylated histone binding proteins (containing a chromodomain, tudor domain, plant homeodomain finger, or WD40 repeat domain) to the chromatin [8].
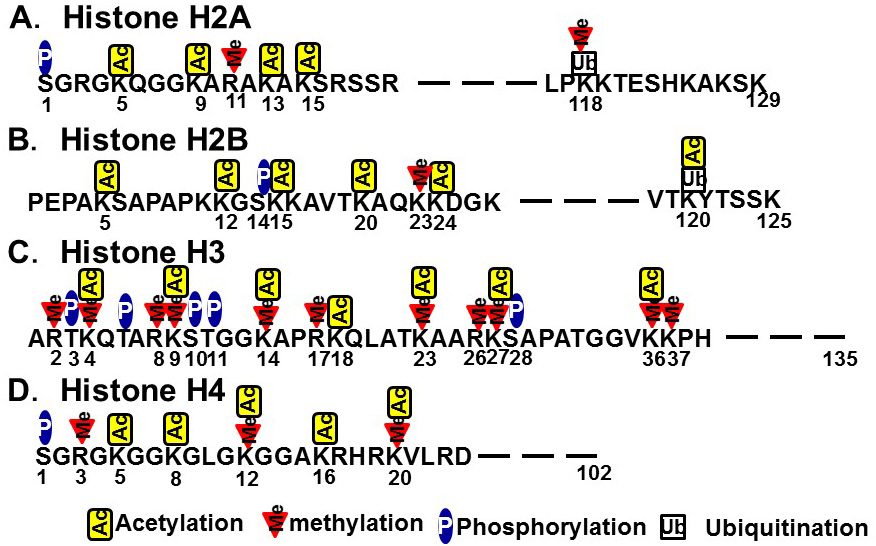
Figure 2 Histone modifications. A) Histone H2A. B) Histone H2B. C) Histone H3. D) Histone H4.
Heterochromatin is formed by enhancing the methylation of CpG islands mediated by DNMT bound to heterochromatin binding proteins on the methylated histones of CpG islands. In contrast, euchromatin is formed by the demethylation of CpG islands and subsequent histone acetylation mediated by HAT [9] (Figure 3). The induction of histone H3K4 monomethylation around the nuclear factor-κB gene by exposing primary human aortic endothelial cells to high glucose for a relatively short time (16 h) was reported to be maintained for 6 days when the cells were incubated under normal glucose conditions [10]. Therefore, it is postulated that environmental factors prevailing during development can lead to a predisposition to diseases later in life that is controlled by epigenetics (Figure 4).
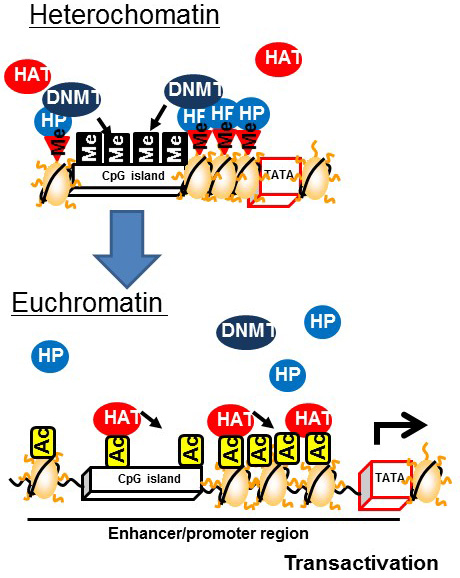
Figure 3 Changes associated with the formation of euchromatin from heterochromatin
Epigenetic Responses to Environmental Factors
It is not fully understood how environmental factors, including nutrient availability during development, affect metabolic gene expression and subsequent development of metabolic diseases. The classic model involves binding of transcription factors to cis-elements located in promoter/enhancer regions. Transcription factor-associated HAT acetylates histones in promoter and enhancer regions, and subsequently, bromodomain proteins bind to the acetylated histones to induce the formation of transcriptional initiation complexes around the TATA box of the promoter region. Nutrients, such as vitamins A and D, as well as unsaturated fatty acids and their metabolites, are ligands for nuclear receptors [retinoic acid receptor, vitamin D receptor, and peroxisome proliferator-activated receptor (PPAR), respectively] [11] (Figure 5A).
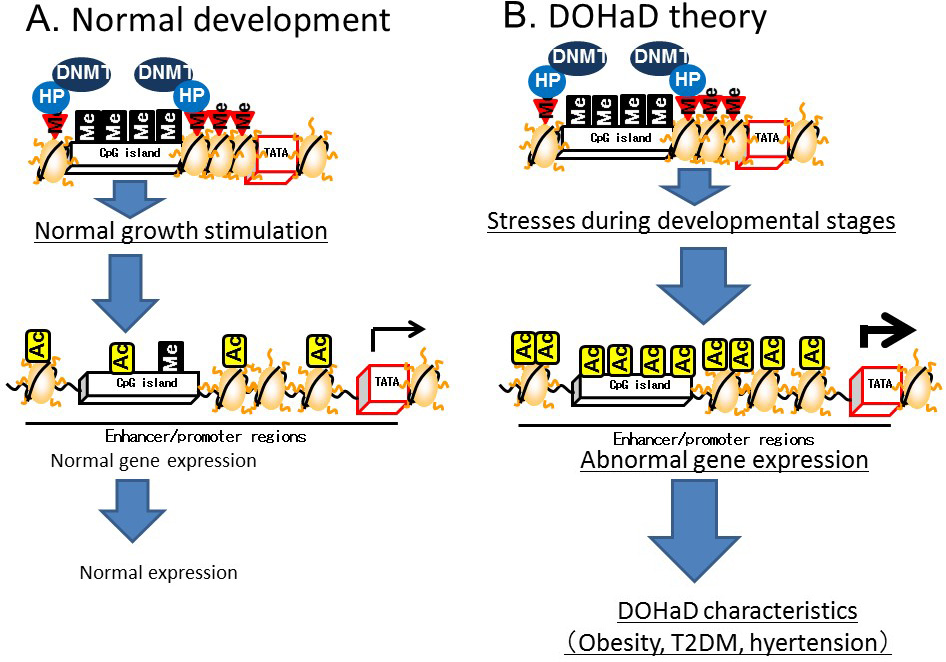
Figure 4 Developmental Origins of Health and Disease (DOHaD) theory based on epigenetics
Recent studies conducted by ourselves and others have demonstrated that histone acetylation around the gene body region is important for regulating the magnitude of transcription. Histone acetylation in the gene body region recruits an acetylated histone binding protein, bromodomain containing 4 (BRD4). This is subsequently bound by positive elongation factor b (P-TEFb), which is a cyclin T1–CDK9 complex. BRD4 and the associated P-TEFb complex on the gene body region enhance transcriptional elongation by phosphorylating RNA polymerase II at the second serine residue of the C-terminal domain [12,13,14] (Figure 5B).
DNA methylation in the gene body regions is positively correlated with transcription [15] and recent studies have demonstrated a relationship with transcriptional elongation. Transcriptional initiation is terminated by CpG islands located in the upstream region by DNMT3a, which is a subtype of DNMT3 (Figure 6A). On the other hand, transcriptional elongation is terminated by methylation of DNA in the gene body region [16]. Histone H3K36 methylation induced by a histone methyltransferase, SET2, represses the transcriptional elongation reaction by reducing histone acetylation via recruiting HDACs on the gene body [17,18,19]. The methylated histone H3K36 recruits another DNMT, DNMT3b, to induce DNA methylation in the gene body region (Figure 6B). Thus, DNMT3a dysfunction leads to the disruption of transcriptional initiation by changing DNA methylation in the promoter/enhancer region, while dysfunctional DNMT3b leads to disrupted transcriptional elongation by methylation of DNA in the gene body region.
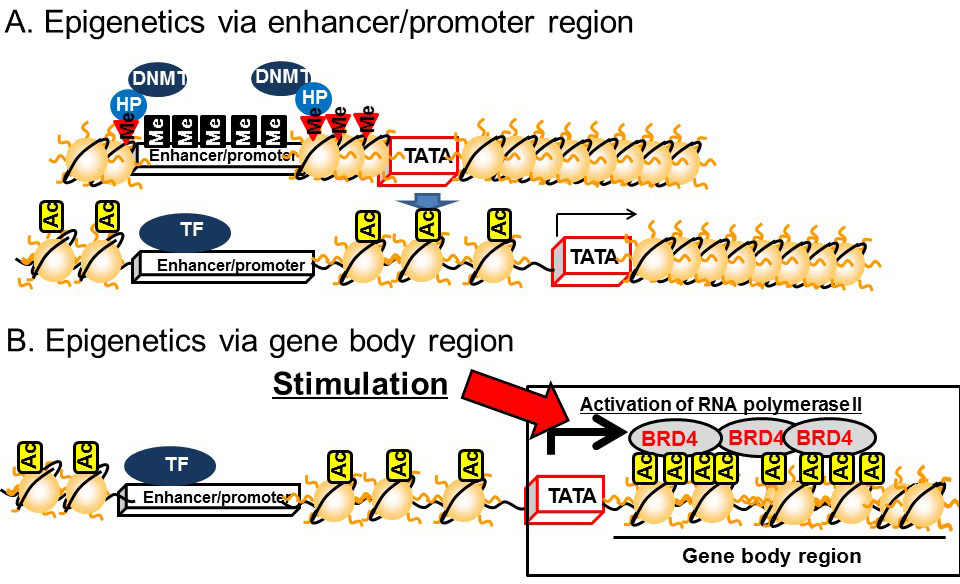
Figure 5 Epigenetics of the enhancer/promoter region and the gene body. A) Epigenetics of the enhancer/promoter region. B) Epigenetics of the gene body.
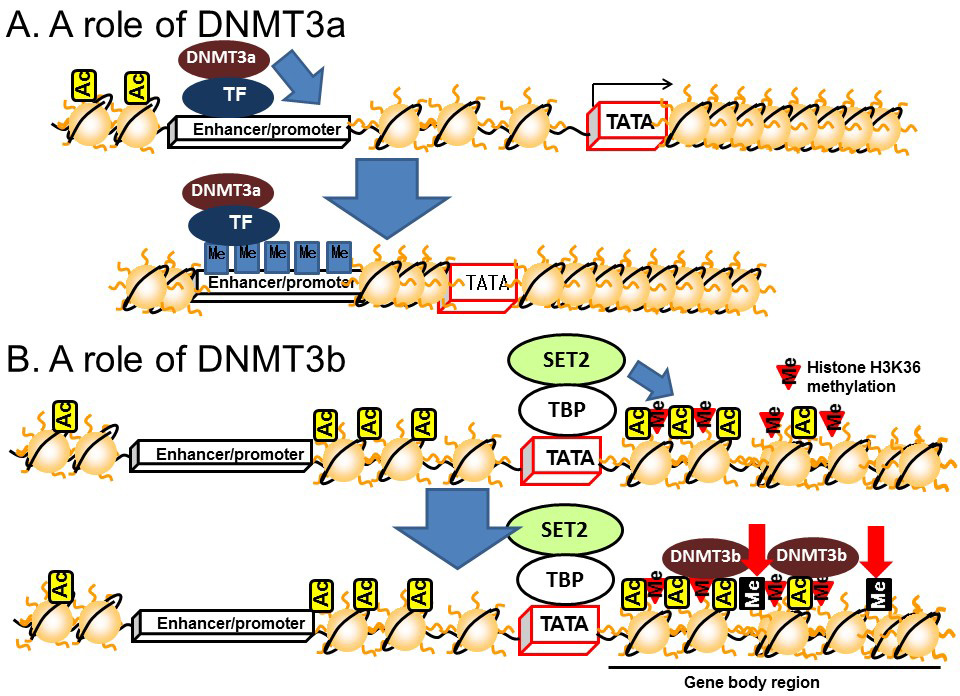
Figure 6 Roles of DNMT3 in the enhancer/promoter and gene body regions. A) DNMT3a. B) DNMT3b
The epigenetic model of gene regulation based on histone modifications and DNA methylation in the gene body region explains the efficacy of mRNA synthesis. Our recent studies in rats have demonstrated that carbohydrate intake increases histone acetylation in the gene body region, rather than in the promoter/enhancer region, of carbohydrate metabolism-related genes, such as sucrose-isomaltase, sodium glucose transporter 1, and glucose transporter 5 in the small intestine [12,13], and fatty acid synthase in the liver during the suckling–weaning transition period [14]. In addition, we have demonstrated that fructose-inducible genes related to fat accumulation, such as Cyp8b1, Dak, and Plin5, are regulated by BRD4 in the liver [15]. Histone acetylation in the gene body region of adiponectin has also been reported to be accompanied by the induction of mRNA synthesis during the differentiation of 3T3-L1 adipocytes [16]. Disturbance of the expression of these genes leads to metabolic diseases and animals exposed to low energy and low protein conditions during the fetal period show abnormal expression of genes regulated by BRD4 and/or histone acetylation in the gene body region; therefore, BRD4-driven gene body acetylation is implicated as an epigenetic mechanism for the development of metabolic diseases induced by the fetal nutritional environment. Indeed, heterozygous Brd4-knockout mice showed growth abnormalities, such as reduced adiposity and abnormal bone metabolism, similar to those of mice exposed to fetal malnutrition [17]. Although there is little evidence supporting a role for the histone H3K36 in this process, our recent study demonstrated that a starvation signal induced H3K36 tri/dimethylation in genes expressed in the small intestine of Xenopus laevis [18].
Recent studies have also demonstrated that gene body epigenetics regulate pre-mRNA splicing. For example, one report described that alternatively spliced exons (ASE) were highly methylated and widely bound to a methylated DNA binding protein MeCP2, and ablation of which resulted in the induction of ASE-skipping in mRNA splicing [19]. However, associations between pre-mRNA spliceosome and DNA methylation or MeCP2 have not yet been established. The relationships between pre-mRNA splicing and gene body epigenetics and between gene body epigenetics and DOHaD require further investigation.
The Mechanism of Epigenetics in the DOHaD Theory
Several animal studies have demonstrated that nutritional conditions during development disrupt epigenetic regulation later in life. For example, the expression of insulin and insulin promoter factor 1 (PDX1), which is a transcription factor for insulin expression in the pancreas, was lower in adult mice exposed to low energy in the fetal period compared with that in mice exposed to normal energy levels. Exposure to starvation conditions in the fetal period induced DNA methylation and histone H3K9 dimethylation, and reduced histone H3 acetylation in the promoter region of Pdx1. Malnutrition during the fetal stage repressed Glut4 gene expression in skeletal muscle, while H3K9 dimethylation, DNMT3a binding, and histone deacetylase (HDAC)1 and 4 were induced and histone H3K14 acetylation in the Glut4 promoter region was reduced [20]. These results suggest that malnutrition during the fetal period reduces insulin activity and induces impaired glucose tolerance in epigenetic disorders of the pancreas and skeletal muscle [20] (Figure 7A).
A low-protein diet fed to mice during pregnancy led to DNA demethylation in the promoter/enhancer regions of Ppara, which encodes a transcription factor for lipid metabolism, in the liver and heart [21], and in the glucocorticoid receptor (Gr) in the heart [22] of their newborn offspring, resulting in increased Ppara and Gr expression.
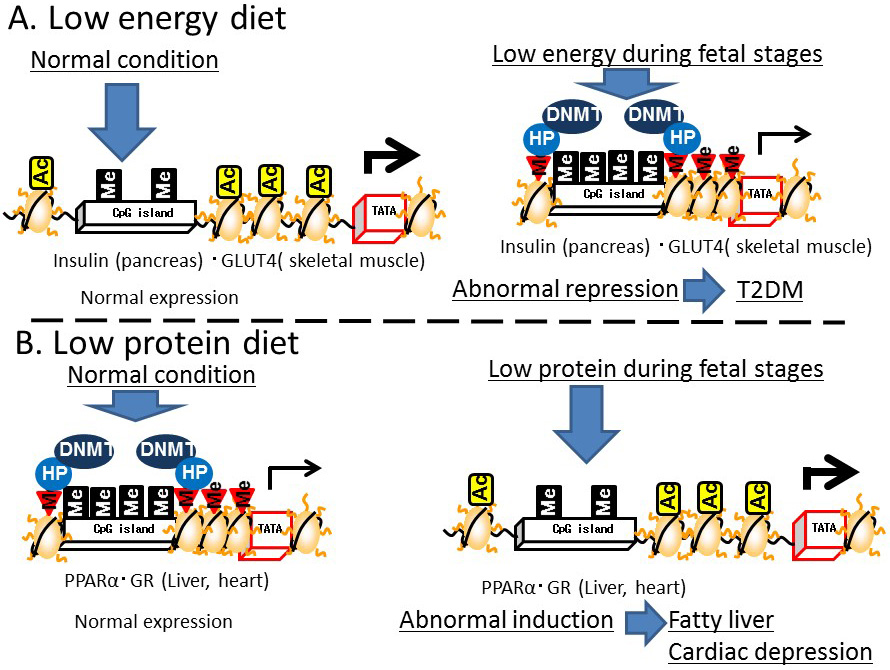
Figure 7 Effects of environmental conditions on Developmental Origins of Health and Disease (DOHaD) epigenetics. A) Low-energy diet. B) Low-protein diet
In addition, a restricted protein diet fed to pregnant rats enhanced the expression of Dnmt1 in the liver of their newborn offspring [23]. A cause of the reduced DNA methylation in the promoter/enhancer regions of Ppara and Gr in the offspring of protein-restricted mothers was reduced Dnmt1 expression. Folic acid supplementation in the pregnant mice led to improved DNA methylation in the upstream region of these genes in the offspring. However, Dnmt3 expression in the newborns was not affected by exposure of the pregnant mice to a protein-restricted diet. DNMT1 maintains DNA methylation by attaching the methyl group to the complementary strand of previously hemi-methylated DNA during replication, while DNMT3 is responsible for de novo methylation. Thus, protein restriction may lead to disruption of the maintenance of DNA methylation during cell division in a pregnancy. In addition, deficiency of choline, which is a nutrient associated with one-carbon and methyl-group metabolism, was shown to induce DNA methylation throughout the genome and around the insulin-like growth factor 2 gene (Igf2), accompanied by the DNA methylation around Dnmt1 [24]. These results indicate that protein-restriction and deficiency of folic acid and choline during the fetal period induce epigenetic disruption in adulthood. Subjecting pregnant rats to protein-restriction led to fatty livers in the offspring [25]. PPARA enhances the utilization of fat under conditions of reduced carbohydrate metabolism, such as insulin-resistance, while GR induces gluconeogenesis and fat accumulation. Excessive PPARA expression in the heart is associated with cardiac muscle disorders [26]. Therefore, the induction of PPARA and GR expression in the heart and liver triggered by a reduction in DNA methylation around the genes encoding these proteins could lead to cardiac muscle disorders and fatty liver (Figure 7B). Regarding gene body epigenetics, DNA methylation in the gene body of liver X receptor A (Lxra) was reduced in the liver of F1 and F2 intrauterine growth-restricted mice [27]. However, since most animal model studies focused on the epigenetics of promoter/enhancer regions, further studies are required to establish the relationship between the epigenetics of gene body regions and developmental programming/DOHaD theory.
The relationship between genomic imprinting and DOHaD is still unclear. However, the fetal environment has been confirmed to induce alterations in the expression and promoter DNA methylation of the paternally-imprinted IGF2 gene in humans and animals. Furthermore, expression of the paternally-inherited IGF2 gene is enhanced by DNA methylation of the differentially methylated region (DMR) located upstream of the adjacent H19 gene, which facilitates the access of proteins to an enhancer region. In contrast, in the case of the maternally-IGF2 gene, the DMR is demethylated and binding of the CTCF protein to this region inhibits the access of proteins to an enhancer region and inactivates the IGF2 gene [28]. It has been reported that the DNA methylation of the DMR upstream of paternally-inherited H19 and IGF2 gene expression are lower in patients with Silver–Russell syndrome [29,30]. Therefore, the fetal environment could affect genomic imprinting, particularly that of the IGF2 gene, and alterations in genomic imprinting may affect the development of metabolic disorders and psychiatric disorders in newborns. However, this issue remains to be clarified in further studies.
Regarding psychiatric disorders, symptoms of schizophrenia, including hyperkinetic disorder, have been reported in the offspring of rats subjected to immobilization stress during pregnancy. Specifically, following the induction of DNA methylation in the promoter regions of the reelin and glutamate decarboxylase 1 genes, reduction of their expression and the induction of Dnmt1 gene expression in the frontal cortex were observed in the offspring of rats subjected to immobilization stress during pregnancy [31]. This indicates that the development of psychiatric disorders which are induced by the fetal environment are caused by epigenetic defects.
Epigenetic Mechanisms Associated with Metabolic Disease According to the DOHaD Theory
In humans, it is not yet known whether the development of metabolic and psychiatric disorders induced by the fetal environment is regulated by epigenetic memory. A cohort study conducted in subjects born to mothers who had suffered malnutrition in the Netherlands during the Second World War showed that DNA methylation in the promoter region of IGF2 was significantly lower in the blood of subjects exposed to these conditions during early gestation compared with that detected in their siblings who had not [4]. Methylation of the IGF2 promoter region was also found to be lower in lymphocytes isolated from umbilical cord blood of infants born to subjects with gestational diabetes than in those born to healthy subjects [32]. In addition, DNA demethylation of the IGF2 promoter region in these cells was positively associated paternal obesity [33]. However, the roles of IGF2 in the development of metabolic diseases are still unclear. An animal study showed that a high-fat diet fed to the offspring of rats exposed to a low-protein diet during pregnancy not only induced obesity and insulin resistance, but also upregulated expression of the Igf2 gene [34]. Therefore, it can be speculated that exposure to environmental factors during human pregnancy induces metabolic and psychiatric disorders by altering the methylation of genes, including IGF2. It has been reported that carbohydrate restriction during pregnancy is positively associated with methylation in the promoter region of retinoid X receptor A (RXRA) in lymphocytes isolated from umbilical cord blood [35]. In addition, the body mass index (BMI) of pregnant women is positively associated with methylation of the PPARγ co-activator 1α (PGC1A) promoter in their newborns [36]. Methylation of the aryl-hydrocarbon receptor DNA promoter in newborns was also reported to be positively associated with maternal obesity during pregnancy, infant gestational age, and birth weight for gestational age [37]. However, there is no evidence in humans that environmental factors during pregnancy alter the methylation of the GLUT4, PPARA, GR, and LXR genes that are affected in animal models. Despite the absence of reports of gene body epigenetic markers associated with metabolic disease development in relation to the developmental programming/DOHaD theory, several studies have been conducted focusing on promoter/enhancer regions. For example, methylation of the gene body region of the glucokinase (GCK) gene in peripheral blood samples was lower in subjects with coronary heart disease and higher in subjects with essential hypertension compared with that in healthy subjects [38,39]. A minor polymorphism in DNMT3B, which regulates DNA methylation in gene body regions, was also associated with an increased risk of premature birth [40]. Thus, it is possible that disrupted gene body epigenetics is linked to developmental programming/DOHaD theory, although this hypothesis requires further examination. In addition, the relationship between histone modification and DNA methylation induced by the fetal environment has not been studied in humans. Further studies using peripheral and umbilical cord blood are required to clarify these relationships in humans.
Conclusion
In this review, we discuss two models of epigenetics based on the promoter/enhancer region and the gene body region. Several studies have demonstrated the existence of epigenetic memory in promoter/enhancer regions and in gene body regions in animals and humans exposed to environmental stress conditions, including malnutrition, during the fetal period as well as in adulthood. However, these findings are preliminary and further studies are required to elucidate the epigenetic mechanisms underlying the development of metabolic diseases associated with exposure to environmental stress during development.
Competing Interests
The authors have declared that no competing interests exist.
Acknowledgments
We thank Dr. Ozato Keiko from the National Institute of Health for providing the chance to study BRD4. We thank Tom Buckle, MSc, from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Funding
Our work cited in this review was supported by Grants-in-Aid for Young Scientists (22680054) and for Scientific Research (26282023, 17H01964) from the Ministry of Education, Culture, Sports, Science and Technology, Japan (MEXT), the Takeda Science Foundation and the Uehara Memorial Foundation.
References
- Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ. Weight in infancy and death from ischaemic heart disease. Lancet. 1989;2(8663):577-80. [CrossRef]
- Harder T, Rodekamp E, Schellong K, Dudenhausen JW, Plagemann A. Birth weight and subsequent risk of type 2 diabetes: a meta-analysis. American journal of epidemiology. 2007;165(8):849-57. [CrossRef]
- Barker DJ. The fetal and infant origins of adult disease. BMJ. 1990;301(6761):1111. [CrossRef]
- Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A. 2008;105(44):17046-9. [CrossRef]
- St Clair D, Xu M, Wang P, Yu Y, Fang Y, Zhang F, et al. Rates of adult schizophrenia following prenatal exposure to the Chinese famine of 1959-1961. Jama. 2005;294(5):557-62. [CrossRef]
- Gluckman PD, Seng CY, Fukuoka H, Beedle AS, Hanson MA. Low birthweight and subsequent obesity in Japan. Lancet. 2007;369(9567):1081-2. [CrossRef]
- Gluckman PD, Hanson MA. The Developmental Origins of Health and Disease. Basic & Clinical Pharmacology & Toxicology. 2008;102(2):90.
- Bruce Alberts AJ, Julian Lewis, David Morgan, Martin Raff, Keith Roberts, Peter Walter. Molecular Biology of the Cell, 6th ed, 2015.
- Persson J, Ekwall K. Chd1 remodelers maintain open chromatin and regulate the epigenetics of differentiation. Experimental cell research. 2010;316(8):1316-23. [CrossRef]
- El-Osta A, Brasacchio D, Yao D, Pocai A, Jones PL, Roeder RG, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. The Journal of experimental medicine. 2008;205(10):2409-17. [CrossRef]
- Xu L, Glass CK, Rosenfeld MG. Coactivator and corepressor complexes in nuclear receptor function. Curr Opin Genet Dev. 1999;9(2):140-7. [CrossRef]
- Inoue S, Mochizuki K, Goda T. Jejunal induction of SI and SGLT1 genes in rats by high-starch/low-fat diet is associated with histone acetylation and binding of GCN5 on the genes. J Nutr Sci Vitaminol (Tokyo). 2011;57(2):162-9. [CrossRef]
- Honma K, Mochizuki K, Goda T. Induction by fructose force-feeding of histone H3 and H4 acetylation at their lysine residues around the Slc2a5 gene and its expression in mice. Biosci Biotechnol Biochem. 2013;77(11):2188-91. [CrossRef]
- Morishita S, Mochizuki K, Goda T. Bindings of ChREBP and SREBP1, and histone acetylation around the rat liver fatty acid synthase gene are associated with induction of the gene during the suckling-weaning transition. J Nutr Sci Vitaminol (Tokyo). 2014;60(2):94-100. [CrossRef]
- Yamada A, Honma K, Mochizuki K, Goda T. BRD4 regulates fructose-inducible lipid accumulation-related genes in the mouse liver. Metabolism. 2016;65(10):1478-88. [CrossRef]
- Sakurai N, Mochizuki K, Goda T. Modifications of histone H3 at lysine 9 on the adiponectin gene in 3T3-L1 adipocytes. J Nutr Sci Vitaminol (Tokyo). 2009;55(2):131-8. [CrossRef]
- Houzelstein D, Bullock SL, Lynch DE, Grigorieva EF, Wilson VA, Beddington RS. Growth and early postimplantation defects in mice deficient for the bromodomain-containing protein Brd4. Molecular and cellular biology. 2002;22(11):3794-802. [CrossRef]
- Tamaoki K, Okada R, Ishihara A, Shiojiri N, Mochizuki K, Goda T, et al. Morphological, biochemical, transcriptional and epigenetic responses to fasting and refeeding in intestine of Xenopus laevis. Cell & bioscience. 2016;6:2. [CrossRef]
- Maunakea AK, Chepelev I, Cui K, Zhao K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell research. 2013;23(11):1256-69. [CrossRef]
- Pinney SE, Simmons RA. Epigenetic mechanisms in the development of type 2 diabetes. Trends in endocrinology and metabolism: TEM. 2010;21(4):223-9. [CrossRef]
- Lillycrop KA, Phillips ES, Jackson AA, Hanson MA, Burdge GC. Dietary protein restriction of pregnant rats induces and folic acid supplementation prevents epigenetic modification of hepatic gene expression in the offspring. The Journal of nutrition. 2005;135(6):1382-6.
- Burdge GC, Hanson MA, Slater-Jefferies JL, Lillycrop KA. Epigenetic regulation of transcription: a mechanism for inducing variations in phenotype (fetal programming) by differences in nutrition during early life? The British journal of nutrition. 2007;97(6):1036-46. [CrossRef]
- Lillycrop KA, Slater-Jefferies JL, Hanson MA, Godfrey KM, Jackson AA, Burdge GC. Induction of altered epigenetic regulation of the hepatic glucocorticoid receptor in the offspring of rats fed a protein-restricted diet during pregnancy suggests that reduced DNA methyltransferase-1 expression is involved in impaired DNA methylation and changes in histone modifications. The British journal of nutrition. 2007;97(6):1064-73. [CrossRef]
- Kovacheva VP, Mellott TJ, Davison JM, Wagner N, Lopez-Coviella I, Schnitzler AC, et al. Gestational choline deficiency causes global and Igf2 gene DNA hypermethylation by up-regulation of Dnmt1 expression. The Journal of biological chemistry. 2007;282(43):31777-88. [CrossRef]
- Erhuma A, Salter AM, Sculley DV, Langley-Evans SC, Bennett AJ. Prenatal exposure to a low-protein diet programs disordered regulation of lipid metabolism in the aging rat. American journal of physiology Endocrinology and metabolism. 2007;292(6):E1702-14. [CrossRef]
- Pruimboom-Brees I, Haghpassand M, Royer L, Brees D, Aldinger C, Reagan W, et al. A critical role for peroxisomal proliferator-activated receptor-alpha nuclear receptors in the development of cardiomyocyte degeneration and necrosis. The American journal of pathology. 2006;169(3):750-60. [CrossRef]
- Martinez D, Pentinat T, Ribo S, Daviaud C, Bloks VW, Cebria J, et al. In utero undernutrition in male mice programs liver lipid metabolism in the second-generation offspring involving altered Lxra DNA methylation. Cell metabolism. 2014;19(6):941-51. [CrossRef]
- Renfree MB, Suzuki S, Kaneko-Ishino T. The origin and evolution of genomic imprinting and viviparity in mammals. Philosophical transactions of the Royal Society of London Series B, Biological sciences. 2013;368(1609):20120151. [CrossRef]
- Gicquel C, Rossignol S, Cabrol S, Houang M, Steunou V, Barbu V, et al. Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver-Russell syndrome. Nature genetics. 2005;37(9):1003-7. [CrossRef]
- Yamazawa K, Kagami M, Nagai T, Kondoh T, Onigata K, Maeyama K, et al. Molecular and clinical findings and their correlations in Silver-Russell syndrome: implications for a positive role of IGF2 in growth determination and differential imprinting regulation of the IGF2-H19 domain in bodies and placentas. J Mol Med (Berl). 2008;86(10):1171-81. [CrossRef]
- Matrisciano F, Tueting P, Dalal I, Kadriu B, Grayson DR, Davis JM, et al. Epigenetic modifications of GABAergic interneurons are associated with the schizophrenia-like phenotype induced by prenatal stress in mice. Neuropharmacology. 2013;68:184-94. [CrossRef]
- Chen D, Zhang A, Fang M, Fang R, Ge J, Jiang Y, et al. Increased methylation at differentially methylated region of GNAS in infants born to gestational diabetes. BMC medical genetics. 2014;15:108. [CrossRef]
- Soubry A, Schildkraut JM, Murtha A, Wang F, Huang Z, Bernal A, et al. Paternal obesity is associated with IGF2 hypomethylation in newborns: results from a Newborn Epigenetics Study (NEST) cohort. BMC medicine. 2013;11:29. [CrossRef]
- Claycombe KJ, Uthus EO, Roemmich JN, Johnson LK, Johnson WT. Prenatal low-protein and postnatal high-fat diets induce rapid adipose tissue growth by inducing Igf2 expression in Sprague Dawley rat offspring. The Journal of nutrition. 2013;143(10):1533-9. [CrossRef]
- Godfrey KM, Sheppard A, Gluckman PD, Lillycrop KA, Burdge GC, McLean C, et al. Epigenetic gene promoter methylation at birth is associated with child's later adiposity. Diabetes. 2011;60(5):1528-34. [CrossRef]
- Gemma C, Sookoian S, Alvarinas J, Garcia SI, Quintana L, Kanevsky D, et al. Maternal pregestational BMI is associated with methylation of the PPARGC1A promoter in newborns. Obesity (Silver Spring). 2009;17(5):1032-9. [CrossRef]
- Burris HH, Baccarelli AA, Byun HM, Cantoral A, Just AC, Pantic I, et al. Offspring DNA methylation of the aryl-hydrocarbon receptor repressor gene is associated with maternal BMI, gestational age, and birth weight. Epigenetics. 2015;10(10):913-21. [CrossRef]
- Xu L, Zheng D, Wang L, Jiang D, Liu H, Liao Q, et al. GCK gene-body hypomethylation is associated with the risk of coronary heart disease. BioMed research international. 2014;2014:151723. [CrossRef]
- Fan R, Wang WJ, Zhong QL, Duan SW, Xu XT, Hao LM, et al. Aberrant methylation of the GCK gene body is associated with the risk of essential hypertension. Molecular medicine reports. 2015;12(2):2390-4. [CrossRef]
- Haggarty P, Hoad G, Horgan GW, Campbell DM. DNA methyltransferase candidate polymorphisms, imprinting methylation, and birth outcome. PloS one. 2013;8(7):e68896. [CrossRef]

