

Application of Clinical Next Generation Sequencing in Intensive Care Facilitates Rapid Diagnosis of Neonates with Rare Genetic Disorders
Tobias Geis 1 ![]() , Ute Hehr 2
, Ute Hehr 2![]() , Roland Brandl 3
, Roland Brandl 3![]() , Saskia Herbst 2
, Saskia Herbst 2![]() , Hugo Segerer 4
, Hugo Segerer 4![]() , Michael Melter 4
, Michael Melter 4![]() , Sophie Hinreiner 2
, Sophie Hinreiner 2![]()
- Department of Pediatric Neurology, University Children´s Hospital Regensburg (KUNO), Klinik St. Hedwig, Steinmetzstr.1-3, Regensburg, Germany
- Center for Human Genetics, Luitpoldstr.4, Regensburg, Germany
- Clinic of Radiology, Neuroradiology and Nuclear Medicine, Department of Pediatric Radiology, Klinik St. Hedwig, Steinmetzstr.1-3, Regensburg, Germany
- University Children´s Hospital Regensburg (KUNO), Klinik St. Hedwig, Steinmetzstr. 1-3, Regensburg, Germany
* Correspondence: Tobias Geis ![]() ; Tel.: +49 941 36998; Fax: +49 941 3695424
; Tel.: +49 941 36998; Fax: +49 941 3695424
Received: August 31, 2017 | Accepted: January 29, 2018 | Published: March 06, 2018
OBM Genetics 2018, Volume 2, Issue 1 doi:10.21926/obm.genet.1801015
Academic Editors: Ute Moog and Domenico Coviello
Special Issue: Next Generation Sequencing
Recommended citation: Geis T, Hehr U, Brandl R, Herbst S, Segerer H, Melter M, Hinreiner S. Application of Clinical Next Generation Sequencing in Intensive Care Facilitates Rapid Diagnosis of Neonates with Rare Genetic Disorders. OBM Genetics 2018;2(1):015; doi:10.21926/obm.genet.1801015.
© 2018 by the authors. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
Abstract
Background: Neonatal muscular hypotonia is a common clinical feature on neonatal intensive care units with a broad spectrum of etiologies. Besides more common and obvious underlying conditions like prematurity or Down syndrome many rare disorders with often poor prognosis need to be considered. Congenital brain malformations detected on ultrasound or/and magnetic resonance imaging might constitute the clinical hallmark in differential diagnosis. We report on our approach combining cerebral imaging and application of targeted next generation panel sequencing. Methods: Retrospective review and evaluation of patients´ files and cerebral images of 4 severely affected neonates treated at the neonatal intensive care unit of our university children’s hospital in 2016 with confirmed genetic diagnosis using clinically targeted multi gene panel sequencing and/or Sanger sequencing. Results: Detailed presentation of the phenotype and genotype of 4 neonates with muscular hypotonia and abnormal MR imaging due to genetically confirmed Aicardi-Goutières syndrome (TREX1), Zellweger spectrum disorder (PEX13), Pontocerebellar Hypoplasia (TSEN54) and X-linked Hydrocephalus (L1CAM), respectively. Overview of genetic and metabolic disorders with brain malformations presenting with neonatal onset muscular hypotonia as a leading symptom. Conclusions: Comprehensive multidisciplinary clinical evaluation of patients with neonatal muscular hypotonia allows phenotype based individual genetic testing including novel targeted multi gene panel testing. This approach might ultimately improve the turn-around time and diagnostic yield including a wide range of very rare genetic disorders.
Keywords
Next generation sequencing NGS; gene panel; rare diseases; neonatal; muscular hypotonia; brain malformation; magnetic resonance imaging; floppy; differential diagnosis
1. Introduction
Muscular hypotonia is a common clinical feature seen in neonatal intensive care units (NICU). The very broad spectrum of etiologies with different long-term courses and variable prognoses represents an enormous challenge to the affected families, as well as for the neonatologists and other medical experts involved in the diagnostic process and medical care.
A thorough prenatal, perinatal and family history as well as a comprehensive physical examination to distinguish between central and peripheral hypotonia, with or without muscle weakness, and with or without dysmorphic features, and congenital malformations, respectively, is considered the essential first step in the differential diagnosis [1]. Several diagnostic algorithms for neonatal hypotonia have been proposed [2,3]; however, this might take weeks to months to complete and the period of uncertainty is often perceived as an additional burden for the parents. Furthermore, this time interval may also critically delay disease-specific therapeutic decisions [3], including the parental consent for palliative care for newborns with confirmed life-limiting conditions and consequently, discontinuation of stressful and invasive interventions.
Congenital brain malformations caused by distinct genetic, chromosomal, metabolic or infectious reasons account for a substantial proportion of neonatal hypotonia [1,4]. The early identification of congenital brain malformations in these newborns may provide critical clues for the differential diagnosis and in guiding disease-specific genetic testing. However, genetic testing by conventional Sanger sequencing is limited to the analysis of genes with a major contribution to the phenotype, which often results in the failure to detect causal mutations in an ever-growing list of genes with more minor contributions. More recently, next generation sequencing (NGS) has been applied successfully to the clinical workup of pediatric disorders as a cost-effective tool, and has proven to be pivotal in the goal to shorten and simplify the diagnostic process and ultimately, to improve the clinical outcome of patients [5,6,7].
Here, we report the clinical application of an individual, cerebral MR imaging-based genetic testing strategy, including targeted multigene panel sequencing, for the rapid genetic diagnosis of severe and rare disorders in four critically ill hypotonic neonates.
2. Patients and Methods
2.1. Patients
Four neonates with muscular hypotonia who were treated regularly in the NICU of Regensburg University Children’s Hospital were included in this study. In addition to standard diagnostic measures obtained according to the hospital standard operating procedures, these four neonates underwent neuropediatric and ophthalmologic examinations, cerebral imaging by both sonography and magnetic resonance imaging (MRI) on clinical grounds. For data assessment, the patients’ medical records were reviewed retrospectively and the MR images were re-evaluated by a pediatric radiologist (RB), a pediatric neurologist (TG) and a human geneticist (UH). TG and UH also assessed the newborns and met with their parents directly at the NICU. For genetic testing, blood samples were collected from each patient and analyzed at Center for Human Genetics Regensburg with the informed consent of the parents.
2.2. Molecular genetics
For multigene panel sequencing, genomic DNA from each patient was processed using the Nextera Rapid Capture Enrichment protocol (Illumina, Inc., San Diego, CA, USA) and library quantification was carried out with on a Bioanalyzer (High Sensitivity DNA Kit; Agilent Technologies, Böblingen, Germany) and a Qubit (dsDNA HS Assay Kit; Life Technologies, Darmstadt, Germany). The whole exome libraries were sequenced on a NextSeqTM500 system (Illumina, Inc.; Patients 2,3) and clinical exome libraries were sequenced on a MiSeqTM system (Illumina, Inc.; Patient 1). All reads were aligned to the human reference genome (UCSC hg 19, NCBI build 37.1) and variant detection was performed with the SeqNextmodul (JSI medical systems) software tool, which was used to filter and categorize all variants that were found in each neonate. Only variants that were covered more than five times and with a frequency below 1% in the general population were selected for further analysis. All potential mutations were screened and categorized as either previously described pathogenic variants (class 5), or novel sequence variants. All novel sequence variants were further evaluated using a variety of bioinformatic programs (MutationTaster-2 [8], PolyPhen-2, SIFT and Alamut-2 (version 2.1; Interactive Biosoftware, Rouen, France). Nucleotide and protein conservation, protein domains and position close to other known mutations were also taken into account as well as population frequencies from the dbSNP database, Ensembl, gnomAD and the Exome Variant Server. After in silico characterization, the sequence variants were classified as class 1–5 variants using a system adapted according to the algorithm suggested by Richards et al. 2014 [9,10]: benign (class 1), likely benign (class 2), uncertain significance (class 3), likely pathogenic (class 4) and pathogenic (class 5).
Sanger sequencing was performed on the ABI 3100Dx XL Avant Sequencer using ABI Prism Big-Dye Terminator Cycle Sequencing Kit version 3.1 (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s recommendations. This method was carried out to confirm sequence variants identified in neonates by multigene panel sequencing and for parental carrier testing as well as for targeted conventional single gene Sanger sequencing of Patient 4.
3. Results
3.1. Patient 1
This male child was born at 37 3/7 weeks of gestation by cesarian section as the first child of healthy non-consanguineous German parents. He had a birth weight of 1,865 g (3.0 SD below the mean), a head circumference of 29.3 cm (3.7 SD below the mean) and Apgar scores of 9 and 10 at 1 and 5 minutes, respectively. The pregnancy was complicated by intrauterine growth retardation, microcephaly and cerebral malformation with hyperechogenic signal alterations detected by prenatal ultrasound at 34 weeks of gestation. Postnatal neuropediatric examination revealed muscular hypotonia and mild bilateral flexion contractures of the elbows, knees and ankles. Feeding by nasogastric tube was necessary due to weak sucking. Laboratory analysis revealed elevated C-reactive protein (maximum 39 mg/l), low platelet count (minimum 35/nl) and elevated GGT (γ-glutamyltransferase; maximum 727 U/l) but the workup did not reveal the presence of infectious diseases, including cytomegalovirus (CMV). Total protein (328 mg/dl) and cell counts (43/µl, lymphocytic) were elevated in the cerebrospinal fluid (CSF). Ophthalmologic fundoscopy revealed a bright fundus with a gray colored optic disk, but no signs of chorioretinitis. Cardiac and abdominal ultrasounds were normal. Cranial ultrasound and cerebral MRI showed extensive calcification of the basal ganglia and the periventricular white matter, dilatation of the ventricular system, a thin corpus callosum and a pachygyria-like cortical disorganization (Figure 1) and Aicardi–Goutières syndrome was suspected on clinical grounds. Due to this pseudo-TORCH clinical presentation with thrombocytopenia and CSF lymphocytosis, an Aicardi–Goutières multigene panel was selected for genetic testing, which started on day 10 of life. The following genes were included in the panel: ADAR, IFIH1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, TREX1. Compound heterozygous TREX1 mutations p.Arg169His (class 5) and p.Cys97* (class 4) were identified on the day 19 of life, confirming the clinical diagnosis of Aicardi–Goutières syndrome Type 1 as a rare disorder with central muscular hypotonia [11]. The TREX1 missense mutation p.Arg196His, also known as p.Arg114His, is a hotspot mutation leading to an altered reference transcript that has been repeatedly observed in independent patients [12]. The boy was discharged from the hospital in a stable condition on day 20 after palliative home care had been organized.
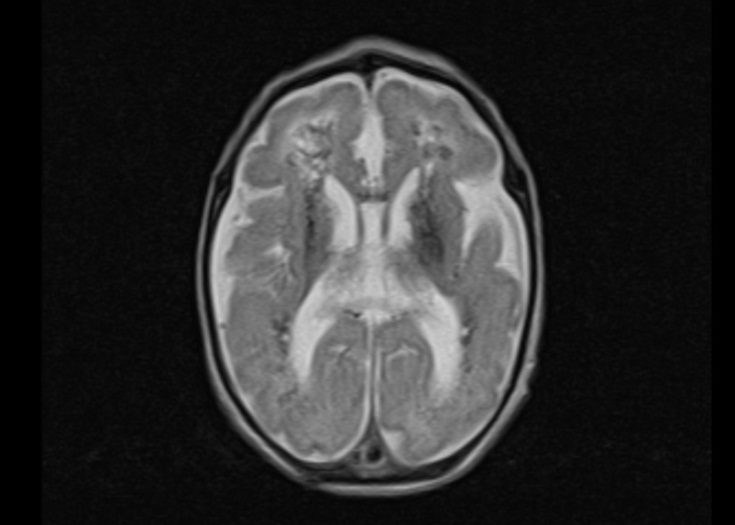
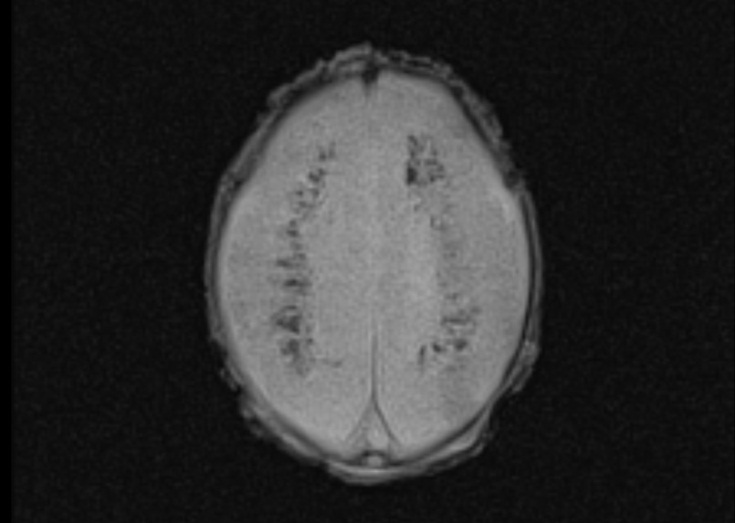
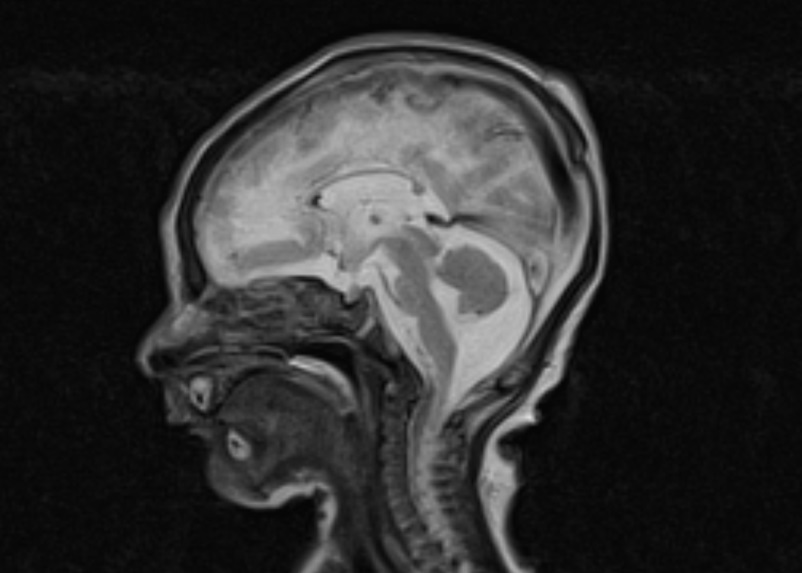
Figure 1 Magnetic resonance images of a male neonate on day 3 of life. The transverse T2-weighted image shows irregular white matter signals in the periventricular regions and a pachygyria-like bilateral cortical disorganization of the frontal area (a). Periventricular hypointense signal abnormalities in the transverse FLASH (fast low angle shot) image suggesting calcification (b). Note the thin corpus callosum in the midsagittal T2-weighted image (c).
3.2. Patient 2
This female patient was the third child of healthy consanguineous parents from Syria. She was born at 38 2/7 weeks of gestation by cesarian section out of greenish stained amniotic fluid. The baby had respiratory problems requiring intratracheal intubation and was admitted to the NICU. Her birth weight was 2,300g (1st centile), length 46 cm (2nd centile), head circumference 34 cm (34th centile) and Apgar scores were 5 and 7 at 1 and 5 minutes, respectively. On day 3 of life she developed epileptic seizures with staring, eye deviation and nystagmus and a pathological seizure pattern in the amplitude integrated electroencephalogram (EEG). Neuropediatric examination revealed profound muscular hypotonia, reduced spontaneous movements and lack of the patellar reflexes; she had dysmorphic facial features with an enlarged anterior fontanel and dysplastic nails on all toes. Laboratory examinations showed elevated C-reactive protein (maximum 15 mg/l) and interleukin-6 (maximum 189 pg/ml) and normal values for liver enzymes, ammonia, bilirubin, blood counts and creatine kinase. No ocular abnormalities were found, but cardiac ultrasound revealed a peri-membranous ventricular septal defect. Cranial ultrasound showed distinct small cysts in the left periventricular region and the cerebral MRI demonstrated a thin corpus callosum and a small vermis in combination with an irregular cortical gyration that was most prominent in the frontal area, suggesting a polymicrogyria-like cortical malformation (Figure 2). In the metabolic workup, the blood concentration of very long chain fatty acids was elevated and the levels of plasmalogens were reduced. This characteristic phenotype prompted targeted genetic expanded panel sequencing for Zellweger spectrum disorders on day 14 of life. The following genes were included in the panel: NPC1, NPC2, PEX1, PEX10, PEX12, PEX13, PEX14, PEX16, PEX19, PEX2, PEX26, PEX3, PEX5, PEX6, SMPD1, OTC. The clinical diagnosis of Zellweger syndrome was confirmed by the observation of a homozygous 14-bp deletion c.107_120del (p.Gly36Aspfs*26; class 4) in the PEX13 gene on day 25 of life [13,14]. The poor prognosis in the absence of any causal treatment was discussed with the parents and palliative supportive care was initiated; after 2 weeks at home the child was readmitted to the NICU with an acute bronchiolitis and died due to respiratory syncytial virus infection at the age of 8 weeks.
3.3. Patient 3
This female patient was born at 41 1/7 weeks of gestation after an uneventful pregnancy. She was the second child of healthy non-consanguineous German parents. Her birth weight was 3,805 g (69th centile), head circumference 34.7 cm (34th centile) and her Apgar scores were 7 and 9 at 1 and 5 minutes, respectively. Due to muscular hypertonia with general hyperexcitability and limb tremors, the baby was admitted to the NICU. She had seizure-like episodes beginning on day 6, with high muscle tone, eye deviation, bilateral limb cloni and cyanosis. The baby was treated for suspected epileptic seizures with intravenous pyridoxine and phenobarbital. Neuropediatric examination revealed initial marked muscular hypertonia with flexion posture of the arms and legs, reduced spontaneous movements and an exaggerated startle response to touch and noise, with clonus-like oscillations of the limbs. Routine laboratory examinations, including metabolic workup, EEG and amplitude integrated EEG were normal. Cranial ultrasound revealed pronounced cerebellar hypoplasia and cerebral MRI confirmed severe hypoplasia of the cerebellum and brainstem (Figure 3). As the clinical course progressed, the hypertonia of the limbs improved and the seizure-like hypertonic attacks disappeared without medical treatment. By the end of her second week of life, the baby became floppy with muscular hypotonia of the trunk and feeding by a nasogastric tube was necessary due to weak suckling. Targeted genetic panel sequencing for the more common genetic forms of pontocerebellar hypoplasia was initiated on day 13. The following genes were included in the panel: B3GALNT1, CASK, DCX, EXOSC3, RARS2, RELN, SEPSECS, TSEN2, TSEN34, TSEN54, VRK1. The clinical diagnosis of a pontocerebellar hypoplasia (PCH) was confirmed by the presence of the homozygous hotspot mutation p.Ala307Ser (class 5: pathogenic variant [15]) in the TSEN54 gene on day 28 of life [16,17]. The child was discharged from the hospital in a stable condition with continued nasogastric feeding at the age of 4 weeks.
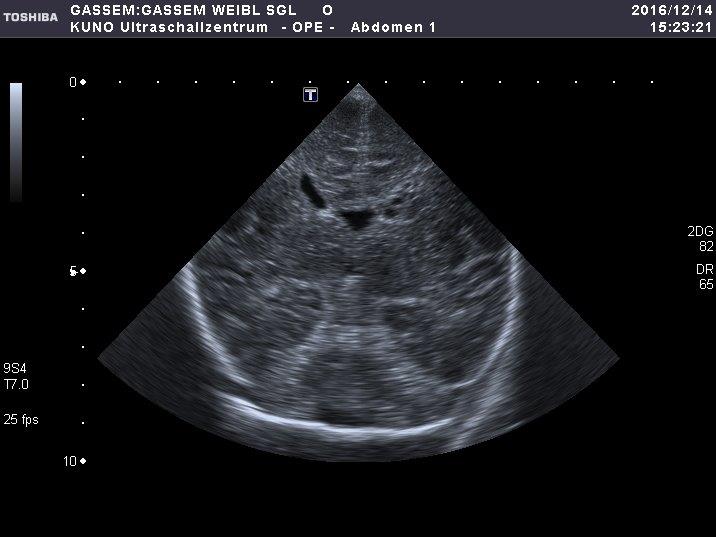
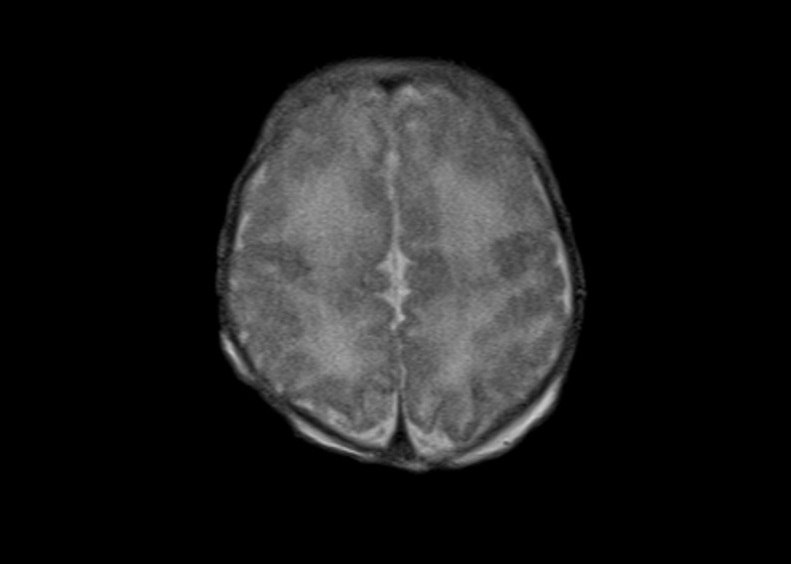
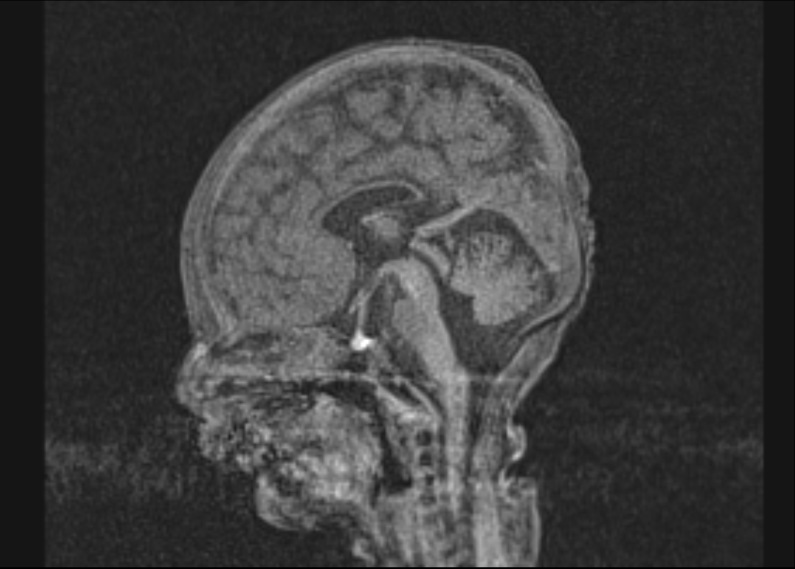
Figure 2 Coronal section of a B-mode ultrasound scan of a female neonate on day 3 of life showing small cysts in the left periventricular region (a). The transverse T2-weighted MR-magnetic resonance image on day 7 of life suggests irregular cortical gyration in the frontal regions (b). Thin corpus callosum in the midsagittal T1 MP-RAGE image (c).
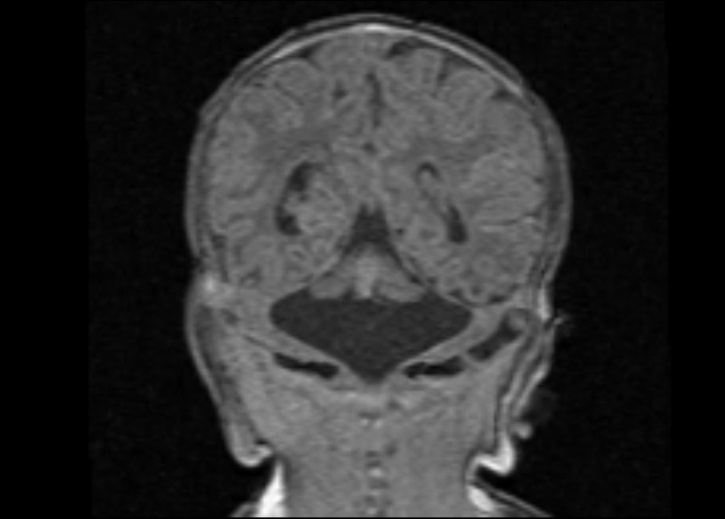
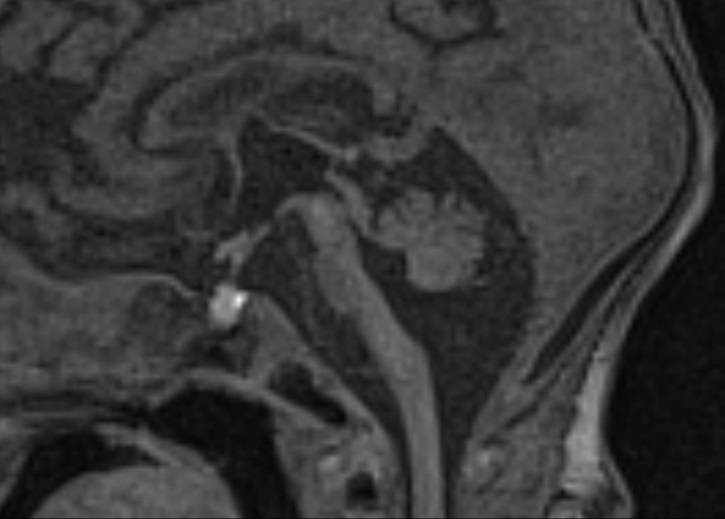
Figure 3 Coronal (a) and midsagittal (b) T1 MP-RAGE images of a female neonate on day 9 of life demonstrating severe hypoplasia of the brainstem and cerebellum with a “dragonfly-like shape” in (a).
3.4. Patient 4
This male patient was born at 36 6/7 weeks of gestation by cesarian section as second child of healthy non-consanguineous German parents. His birth weight was 3,130 g (56th centile), head circumference 39.5 cm (3.4 SD above the mean) and his Apgar scores were 7 and 8 at 1 and 5 minutes, respectively. The pregnancy was complicated by ventricular dilatation detected by prenatal ultrasound at 34 weeks of gestation. Neuropediatric examination revealed muscular hypotonia and ulnar deviation of the wrists, with adducted thumbs. Massive dilatation of the lateral ventricles and the third ventricle was found in the cranial ultrasound, and aqueductal stenosis suspected (Figure 4a, b). On day 2 of life, a ventriculoperitoneal shunt was inserted. After surgery, the patient had irregular breathing with apnea and feeding via a nasogastric tube was required; these symptoms slowly improved as the clinical course progressed. The characteristic combination of hydrocephalus due to aqueduct stenosis together with prenatally suspected agenesis of the corpus callosum and adducted thumbs prompted Sanger sequencing of the L1CAM gene on day 3. The results confirmed the tentative diagnosis of X-linked hydrocephalus due to a hemizygous L1CAM nonsense mutation p.Glu305* (class 4) on day 9 of life [18,19]. The L1CAM mutation was not observed in the mother, indicating the rare event of germline mosaicism; thus, the recurrence risk for further pregnancies is predicted to be only slightly increased when compared to that of the general population. Discussions were held with the parents regarding the poor long-term prognosis of this syndromic form of hydrocephalus, including intellectual disability and progressive spasticity of the lower limbs. The boy was released from the hospital in a stable condition. A cerebral MRI performed to evaluate the shunt control at the age of 10 months showed marked dilatation of the lateral ventricles, severe stenosis of the aqueduct and agenesis of the corpus callosum (Figure 4c, d). The agenesis of the corpus callosum had not been documented on the postnatal ultrasound due to the extensive hydrocephalus. At the age of 20 months the patient was severely hypotonic with a frog-like supine position and impaired head control; he had persistent adducted thumbs and was unable to speak any words.
4. Discussion
The four patients (2 girls, 2 boys) described here exhibited severe and characteristic phenotypes of their respective rare diseases [11,13,16,17,18,19] with a prenatal or neonatal onset. They were admitted to the NICU of our children’s hospital between August 2015 and March 2017. The suspected clinical diagnosis was genetically confirmed on days 9 (Patient 4), 19 (Patient 1), 25 (Patient 2) and 28 (Patient 3) of life, helping to guide the further medical care and therapy of these children.
During the last few decades, the broad introduction of genetic testing into routine diagnostic algorithms has led to tremendous improvements in the genetic confirmation of a clinical diagnosis in patients with muscular hypotonia associated with the more common “rare genetic disorders” like spinal muscular atrophy (SMA). The lessons learned from conventional Sanger sequencing were of great value to medical geneticists dedicated to patient care; however, this information has not been widely recognized beyond the genetics community. Comprehensive pretest clinical characterization of each individual patient should be carried out prior to any diagnostic genetic testing. This clinical characterization benefits from interdisciplinary approaches, including a broad range of medical specialties.
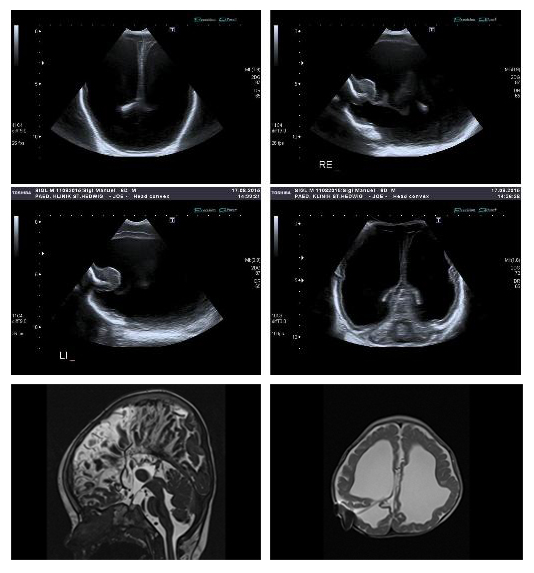
Figure 4 Left lateral sagittal and coronal section of a B-mode ultrasound scan of a male neonate on day 2 of life showing extensive dilatation of the lateral ventricles (a, b). Stenosis of the aqueduct and aplasia of the corpus callosum found in the midsagittal T2 CISS (constructive interference in steady state) image at the age of 10 months. Note the radial orientation of the sulci on the medial hemisphere (c). Transverse T2-weighted image demonstrates severely reduced white matter volume and extensive ventricular dilatation despite inserted ventriculoperitoneal shunt (d).
For the differential diagnosis of neonates with muscular hypotonia, the clinical assessment should commonly include evaluation not only by neonatologists, but also by pediatric neurologists, ophthalmologists, human geneticists and radiologists. In addition, these clinicians should be experienced in the assessment of neonatal cerebral imaging as well as laboratory tests, including evaluation of parameters such as muscle enzymes (e.g. creatine kinase), myasthenic antibodies, metabolic parameters (e.g. lactate), infection and markers of inflammation. The resulting clinical working hypothesis critically guides the type of genetic test chosen to confirm the diagnosis. It is important that ordering physicians are aware that none of the currently available genetic tests is capable of detecting all possible mutation types. More importantly, novel multigene panel sequencing is not suited to the detection of the mutations underlying the more common genetic neuromuscular disorders presenting with severe neonatal hypotonia, which need to be considered first in the differential diagnosis. These disorders include Werdnig–Hofmann disease (congenital spinal muscular atrophy [SMA]), Curschmann–Batten–Steinert syndrome (congenital myotonic dystrophy [DM]) and Prader–Willi syndrome. NGS may indirectly indicate the causal mutations by revealing “coverage gaps” resulting from homozygous exon deletions within the SMN1 gene, which is the most common cause of SMA. However, those exon deletions should be concurrently detected by dose-sensitive methods, such as multiplex ligation-dependent probe amplification (MLPA). This technique is designed to reliably identify deletions and duplications in the SMN1 gene in patients as well as in healthy heterozygous mutation carriers. Large CTG repeat expansions underlying congenital forms of DM can remain entirely undetected by targeted panel sequencing and require repeat-specific detection techniques that are either PCR- and/or hybridization-based. Moreover, detection of mutations in mitochondrial DNA requires specific pre-analytic processing to enrich for mitochondrial DNA in each sample
The observation of structural brain malformations in a neonate with muscular hypotonia adds important information to the differential diagnosis of a variety of disorders including a wide range of rare monogenic conditions. A proper description based on cerebral MRI in the context of the overall clinical presentation often paves the way for targeted genetic testing. Table 1 provides an overview of genetic disorders presenting with neonatal onset of muscular hypotonia as a leading symptom. However, the cost and time-consuming nature of conventional Sanger sequencing has so far limited its application to genes with a major contribution to the respective phenotype, leaving many rare genetic conditions undiagnosed.
The value of targeted multigene panel sequencing clearly lies in the time and cost-effective advantages of simultaneous sequence analysis of a broad range of genes associated with genetically heterogeneous conditions such as pontocerebellar hypoplasia or Zellweger syndrome. This technique offers the possibility of including more genes with a more minor contribution to the phenotype, thus increasing the mutation detection rate and hence, the diagnostic yield. The pretest clinical workup is required to determine whether or not one or more of these other disease-specific, often rapid and less costly assays should be performed in a floppy neonate prior to NGS as a first line diagnostic test to rule out mutations, such as deletions of exons 7 and 8 in the SMN1 gene. Ultimately the multigene panel approach will also provide new insights into more complex modes of inheritance that are not properly considered in current routine diagnostic testing, for instance digenic inheritance, which has recently been demonstrated in some patients with holoprosencephaly [20,21].
5. Conclusion
Here, we present the application of a comprehensive multidisciplinary clinical evaluation of patients with severe neonatal muscular hypotonia at our NICU including a phenotype-based targeted genetic testing approach. This clinical approach including novel NGS multigene panel sequencing allows rapid and cost-effective genetic diagnosis of a variety of conditions including a wide range of very rare genetic disorders. Our data highlight the importance of a thorough pretest clinical workup prior to genetic testing, which may not only critically reduce costs and time to diagnosis, but also supports the classification and interpretation of observed genetic variants in the individual clinical context.
Table 1 Selected genetic and metabolic disorders with brain malformation presenting with neonatal muscular hypotonia as a leading symptom. CC, corpus callosum; BG, basal ganglia; PMG, polymicrogyria; WM, white matter.
|
Disorder |
MRI abnormalities |
Associated core genes |
|
Walker–Warburg syndrome (WWS) Muscle eye brain disease (MEB) Fukuyama muscular dystrophy Other dystroglycanopathies resulting from defective O-glycosylation [22,23] |
Cobblestone lissencephaly, agyria, pachygyria, PMG, hydrocephalus, agenesis or hypoplasia of CC, hypoplastic pons and brainstem, hypoplastic or dysplastic cerebellum, cerebellar cysts, leukencephalopathy |
FKRP, FKTN, ISPD, LARGE, POMGnT1, POMT1, POMT2
|
|
Merosin-deficient muscular dystrophy [4,24]
|
Bilateral cerebral WM abnormality, cerebellar WM abnormality, occipital pachygyria and PMG, hypoplastic cerebellum |
FKRP, LAMA2, LARGE |
|
Zellweger spectrum disorders [13,14]
|
Perisylvian PMG, pachygyria, germinolytic cysts, reduced WM and leukencephalopathy, cerebral atrophy and ventricular dilatation |
PEX1, PEX10, PEX12, PEX13, PEX14, PEX16, PEX19, PEX2, PEX26, PEX3, PEX5, PEX6 |
|
Congenital disorders of glycosylation (CDG) resulting from defective N-glycosylation [4] |
Hypoplastic cerebellum & pons, cerebellar atrophy and hyperintensity |
ALG3, ALG6, ALG8, ALG12, DPM1, MPDU1, MPI, PMM2 |
|
Pontocerebellar hypoplasia (PCH) [17] |
Hypoplastic pons and brainstem, cerebellar hypoplasia, especially of the hemispheres |
CHMP1A, EXOSC3, RARS2, SEPSECS, TSEN2, TSEN34, TSEN54, VRK1 |
|
Lissencephaly spectrum [25]
|
Agyria, pachygyria, subcortical band heterotopia |
PAFAH1B1, DCX, TUBA1A, TUBB2B, DYNC1H1 |
|
Hydrocephalus, agenesis or hypoplasia of CC, aqueductal stenosis |
L1CAM |
|
|
Brain calcifications, especially in BG and WM, reduced WM and leukencephalopathy, cerebral atrophy, deep WM cysts |
ADAR, IFIH1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, TREX1
|
|
|
Smith–Lemli–Opitz syndrome [4,27]
|
Dysgenesis of CC, abnormal septum pellucidum, cerebellar and cerebral atrophy, WM lesions |
DHCR7 |
|
Pyruvate dehydrogenase deficiency [4,28]
|
Dysgenesis of CC, reduced and hyperintense WM, ventricular dilatation I–III, hypoplastic pons |
DLAT, LIAS, PDHA1, PDHB, PDP1, PDX1 |
|
Nonketotic hyperglycinemia (Glycine encephalopathy) [4] |
Hypoplastic or dysgenetic CC, hyperintense signal and reduced diffusion in white matter tracts and BG |
AMT, GCSH, GLDC, |
|
Joubert syndrome [29]
|
Dysplasia of pons and cerebellar vermis & peduncles (molar tooth sign) |
AHI1, C5orf42, CC2D2A, CEP290, TMEM67
|
|
Complex brain malformation with incomplete cleavage of the forebrain; four types: lobar, semilobar, alobar, middle interhemispheric variant |
CDON, DISP1, DLL1, FGF8, FGFR1, GLI2, SHH, SIX3, SUFU, TGIF1, ZIC2 |
|
|
Lowe syndrome [30] |
Ventricular dilatation, periventricular cysts |
OCRL |
|
Wolf–Hirschhorn syndrome [31] |
Ventricular dilatation, reduced and hyperintense cerebral WM, thinning of CC, periventricular cysts |
Array CGH/FISH for the critical region 4p16.3 |
Acknowledgments
Many thanks to Johanna Joe for providing the ultrasound scan shown in Figure 2a.
Funding Source
The authors have no funding resources or grants to declare.
Competing Interests
The authors have no competing interests to declare.
Authors’ Contribution
TG: Main author, correspondance, literature search, proofreading; UH: Interpretaton genetic results, author of "discussion", literature search, proofreading; RB: Interpretation imaging, proofreading; SH: Interpretation genetic results, proofreading; HS: Interpretaton of clinical results, proofreading; MM: Interpretaton of clinical results, proofreading; SH: Interpretation genetic results, author of “methods”, literature search, proofreading.
References
- Prasad AN, Prasad C. The floppy infant: contribution of genetic and metabolic disorders. Brain Dev. 2003; 25: 457-476. [CrossRef]
- Sparks SE. Neonatal hypotonia. Clin Perinatol. 2015; 42: 363-371, ix. [CrossRef]
- Prasad AN, Prasad C. Genetic evaluation of the floppy infant. Semi Fetal Neonatal Med. 2011; 16: 99-108. [CrossRef]
- Whitehead MT, Fricke ST, Gropman AL. Structural brain defects. Clin Perinatol. 2015; 42: 337-361, ix. [CrossRef]
- Stark Z, Tan TY, Chong B, Brett GR, Yap P, Walsh M, et al. A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet Med. 2016; 18: 1090-1096. [CrossRef]
- Vissers L, van Nimwegen KJM, Schieving JH, Kamsteeg EJ, Kleefstra T, Yntema HG, et al. A clinical utility study of exome sequencing versus conventional genetic testing in pediatric neurology. Genet Med. 2017; 19: 1055-1063. [CrossRef]
- Herbst SM, Schirmer S, Posovszky C, Jochum F, Rodl T, Schroeder JA, et al. Taking the next step forward - Diagnosing inherited infantile cholestatic disorders with next generation sequencing. Mol Cell Probes. 2015; 29: 291-298. [CrossRef]
- Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014; 11: 361-362. [CrossRef]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17: 405-424. [CrossRef]
- Matthijs G, Souche E, Alders M, Corveleyn A, Eck S, Feenstra I, et al. Eur J Hum Genet. 2016; 24: 1515. [CrossRef]
- Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A. 2015; 167a: 296-312. [CrossRef]
- Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, et al. Mutations in the gene encoding the 3'-5' DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet. 2006; 38: 917-920. [CrossRef]
- Klouwer FC, Berendse K, Ferdinandusse S, Wanders RJ, Engelen M, Poll-The BT. Zellweger spectrum disorders: clinical overview and management approach. Orphanet J Rare Dis. 2015; 10: 151. [CrossRef]
- Weller S, Rosewich H, Gartner J. Cerebral MRI as a valuable diagnostic tool in Zellweger spectrum patients. J Inherit Metab Dis. 2008; 31: 270-280. [CrossRef]
- Budde BS, Namavar Y, Barth PG, Poll-The BT, Nurnberg G, Becker C, et al. tRNA splicing endonuclease mutations cause pontocerebellar hypoplasia. Nat Genet. 2008; 40: 1113-1118. [CrossRef]
- Maras-Genc H, Uyur-Yalcin E, Rosti RO, Gleeson JG, Kara B. TSEN54 gene-related pontocerebellar hypoplasia type 2 presenting with exaggerated startle response: report of two cases in a family. Turk J Pediatr. 2015; 57: 286-289.
- Rudnik-Schoneborn S, Barth PG, Zerres K. Pontocerebellar hypoplasia. Am J Med Genet C Semin Med Genet. 2014; 166c: 173-183. [CrossRef]
- Weller S, Gartner J. Genetic and clinical aspects of X-linked hydrocephalus (L1 disease): Mutations in the L1CAM gene. Hum Mutat. 2001; 18: 1-12. [CrossRef]
- Fransen E, Lemmon V, Van Camp G, Vits L, Coucke P, Willems PJ. CRASH syndrome: clinical spectrum of corpus callosum hypoplasia, retardation, adducted thumbs, spastic paraparesis and hydrocephalus due to mutations in one single gene, L1. Eur J Hum Genet. 1995; 3: 273-284. [CrossRef]
- Dubourg C, Carre W, Hamdi-Roze H, Mouden C, Roume J, Abdelmajid B, et al. Mutational Spectrum in Holoprosencephaly Shows That FGF is a New Major Signaling Pathway. Hum Mutat. 2016; 37: 1329-1339. [CrossRef]
- Mouden C, Dubourg C, Carre W, Rose S, Quelin C, Akloul L, et al. Complex mode of inheritance in holoprosencephaly revealed by whole exome sequencing. Clin Genet. 2016; 89: 659-668. [CrossRef]
- Godfrey C, Clement E, Mein R, Brockington M, Smith J, Talim B, et al. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain. 2007; 130: 2725-2735. [CrossRef]
- Hehr U, Uyanik G, Gross C, Walter MC, Bohring A, Cohen M, et al. Novel POMGnT1 mutations define broader phenotypic spectrum of muscle-eye-brain disease. Neurogenetics. 2007; 8: 279-288. [CrossRef]
- Leite CC, Lucato LT, Martin MG, Ferreira LG, Resende MB, Carvalho MS, et al. Merosin-deficient congenital muscular dystrophy (CMD): a study of 25 Brazilian patients using MRI. Pediatr Radiol. 2005; 35: 572-579. [CrossRef]
- Di Donato N, Chiari S, Mirzaa GM, Aldinger K, Parrini E, Olds C, et al. Lissencephaly: Expanded imaging and clinical classification. Am J Med Genet A. 2017; 173: 1473-1488. [CrossRef]
- La Piana R, Uggetti C, Roncarolo F, Vanderver A, Olivieri I, Tonduti D, et al. Neuroradiologic patterns and novel imaging findings in Aicardi-Goutieres syndrome. Neurology. 2016; 86: 28-35. [CrossRef]
- Lee RW, Conley SK, Gropman A, Porter FD, Baker EH. Brain magnetic resonance imaging findings in Smith-Lemli-Opitz syndrome. Am J Med Genet A. 2013; 161a: 2407-2419. [CrossRef]
- Ah Mew N, Loewenstein JB, Kadom N, Lichter-Konecki U, Gropman AL, Martin JM, et al. MRI features of 4 female patients with pyruvate dehydrogenase E1 alpha deficiency. Pediatr Neurol. 2011; 45: 57-59. [CrossRef]
- Romani M, Micalizzi A, Valente EM. Joubert syndrome: congenital cerebellar ataxia with the molar tooth. Lancet Neurol. 2013; 12: 894-905. [CrossRef]
- Loi M. Lowe syndrome. Orphanet J Rare Dis. 2006; 1: 16. [CrossRef]
- Righini A, Ciosci R, Selicorni A, Bianchini E, Parazzini C, Zollino M, et al. Brain magnetic resonance imaging in Wolf-Hirschhorn syndrome. Neuropediatrics. 2007; 38: 25-28. [CrossRef]

