

Identification of a Small Supernumerary Marker Chromosome Involving 11p14.1q12.1 in a Prenatal Case: Clinical and Molecular Characterization
Maria Kontodiou 1![]() , Vassilis Paspaliaris 1
, Vassilis Paspaliaris 1![]() , Themistoklis Dagklis 2
, Themistoklis Dagklis 2![]() , Elisavet Siomou 1
, Elisavet Siomou 1![]() , Ahmed Hamid Al-Rikabi 3
, Ahmed Hamid Al-Rikabi 3![]() , Kalliopi Tsita 4
, Kalliopi Tsita 4![]() , Theano Stavroulaki 4
, Theano Stavroulaki 4![]() , Andreas Pampanos 4
, Andreas Pampanos 4![]() , Apostolos Zavlanos 5
, Apostolos Zavlanos 5![]() , Georgios Papaioannou 6
, Georgios Papaioannou 6![]() , Ioannis Papoulidis 1
, Ioannis Papoulidis 1![]() , Loretta Thomaidis 7
, Loretta Thomaidis 7![]() , Emmanouil Manolakos 1,7,8
, Emmanouil Manolakos 1,7,8 ![]()
- Access to Genome P.C., Clinical Laboratory Genetics, Athens-Thessaloniki, Greece
- 3rd Obstetrics and Gynecology clinic, Ippokrateion Hospital, Aristotle University of Thessaloniki, Thessaloniki, Greece
- Jena University Hospital, Friedrich Schiller University, Institute of Human Genetics, Am Klinikum 1, D-07747 Jena, Germany
- Alexandra General Hospital of Athens, Athens, Greece
- 1st Department in Obstetrics & Gynecology AUTh Papageorgiou Hospital, Thessaloniki
- Department of Gynecology, Attikon Hospital, University of Athens, Athens, Greece
- Developmental assessment unit, 2nd department of pediatrics, P. & A. Kyriakou children's hospital, School of medicine, National and Kapodistrian University of Athens, Athens, Greece
- Department of Medical Genetics, University of Cagliari, Binaghi Hospital, Cagliari, Italy
* Correspondence: Emmanouil Manolakos![]()
Received: May 30, 2018 | Accepted: August 15, 2018 | Published: September 18, 2018
OBM Genetics 2018, Volume 2, Issue 3 doi:10.21926/obm.genet.1803035
Academic Editor: Thomas Liehr
Special Issue: Applications of Fluorescence in Situ Hybridization
Recommended citation: Kontodiou M, Paspaliaris V, Dagklis T, Siomou E, Rikabi AHA, Tsita K, Stavroulaki T, Pampanos A, Zavlanos A, Papaioannou G, Papoulidis L, Thomaidis L, Manolakos E. Identification of a Small Supernumerary Marker Chromosome Involving 11p14.1q12.1 in a Prenatal Case: Clinical and Molecular Characterization. OBM Genetics 2018;2(3):035; doi:10.21926/obm.genet.1803035.
© 2018 by the authors. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
Abstract
Background: Small supernumerary marker chromosomes (sSMC) are structurally abnormal chromosomes, and their characterization exclusively by banding cytogenetics is almost impossible. Multicolor fluorescence in situ hybridization approaches for their characterization are effective but expensive and time-consuming. Recently, the application of molecular karyotyping has resulted in improving the characterization of sSMC. Methods: Molecular karyotyping was used for the identification of a sSMC in this study. Results: In this case report, a fetus with a SMC derived from chromosome 11 is described. Conclusions: Molecular karyotyping is suited for sSMC characterization of prenatal cases. The obtained molecular data can directly be used to compare the actual case with data from the literature available on http://ssmc-tl.com/sSMC.html.
Keywords
Chromosome 11; small supernumerary marker chromosomes (sSMCs); chromosomal microarray analysis (CMA).
1. Introduction
Small supernumerary marker chromosomes (sSMCs) are structurally abnormal chromosomes, and they are equal in size or smaller than the chromosome 20 of the same metaphase spread [1]. The incidence of sSMCs has been estimated to be 0.075% - 1.5% in prenatal diagnosis [2]. The heterogeneous group of sSMCs presents a serious genetic counseling problem, especially if they are present de novo, being in a mosaic condition and diagnosed prenatally, as the risk of clinical consequences is not easy to be predicted.
Genetic characterization of an sSMC only by banding cytogenetics is almost impossible. Approaches for its characterization such as sophisticated multicolor fluorescence in situ hybridization (mFISH) approaches or microdissection followed by reverse FISH are effective but time-consuming [3]. FISH based identification of sSMCs shows approximately 50% to be derived from chromosome 15, for which there is a well-established genotype-phenotype correlation [4,5]. However, for most other sSMCs genotype-phenotype correlations are still only rudimentary [5]. Recently, the application of Chromosomal Microarray Analysis (CMA) was introduced in to routine characterization of sSMCs.
Here, a case of an sSMC is described, which was identified prenatally due to advanced maternal age. It turned out to be derived from chromosome 11 and it is compared with previously reported cases from literature.
2. Materials and Methods
Fetal DNA was extracted directly using the Instagene Matrix resin (Bio-Rad Laboratories). Quantitative fluorescent polymerase chain reaction analysis was performed using polymorphic short tandem repeat markers specific for chromosomes 13, 18, 21, X and Y. The PCR fragments were separated in an 8% denaturing polyacrylamide gel electrophoresis on an ALFexpress II DNA sequencer (Amersham Parmacia Biotech) and analyzed using Fragment Analysis software (Amersham).
CMA was carried out on DNA extracted from fresh amniotic fluid of 15 weeks of gestation (w.o.g.) according to standard procedures. CMA was carried out using oligonucleotide aCGH platforms (Agilent technologies, Santa Clara, CA) using an 80 Kb resolution array (kit 60k) as described elsewhere [6]. The reference DNA was of male gender. The statistical test used as a parameter to estimate the number of copies was ADM-2 (provided by the DNA analytics software, Agilent Technologies) with a window of 0.5 Mb and a threshold of 6. Only those copy number changes that affected at least 5 consecutive probes with identically oriented change were considered as copy number variations. Consequently, for most of the genome, the average resolution of this analysis was 200 kb. The position of oligomers refers to the human genome assembly February 2009 (GRCh37/hg19). The Decipher (http://decipher.sanger.ac.uk/application) and Ecaruca (http://agserver01.azn.nl:8080/ecaruca.jsp) databases were used as additional resources. The study was performed with respect to the ethical standards of the Declaration of Helsinki, as revised in 2008.
3. Results
A 39-year-old female was referred for prenatal diagnosis due to advanced maternal age. The family history was unremarkable. Fetal ultrasound examinations at 11, 13 and 15 w.o.g. showed no abnormalities. Nuchal translucency at 13 w.o.g. was 1.9 mm with a calculated Down syndrome risk of 1/1,150.
Chorionic villus sampling (CVS) was performed after parental request due to maternal anxiety at 13 w.o.g. Normal disomic results were obtained for chromosomes 13, 18, 21 and X in quantitative fluorescent polymerase chain reaction test (results not shown).
After cell culture chromosomes prepared from CVS revealed in GTG banding (300 – 400 bands) an sSMC being present in two independent cultures [fetal karyotype according to ISNC 2016: 47,XY, +mar/46,XY [7]]. Parental peripheral blood samples analyses revealed normal karyotypes.
At 15 w.o.g. an amniocentesis was performed and the previous result was confirmed: the fetal karyotype was 47, XY, +mar (dn)/46,XY (Figure 1).

Figure 1 GTG banding of metaphase chromosomes from the amniotic fluid of the fetus.
To characterize the origin and the genetic content of the de novo sSMC, CMA was carried out using DNA derived from fresh amnion sample. A 25.85 Mb duplication chromosome 11 spanning 11p14 to 11q12.1 (positions 30,796,545 to 56,649,983) was identified (Figure 2). According to http://ssmc-tl.com/sSMC.html (Accessed: 24/06/2018) the de novo sSMC [11] spans the critical region of sSMCs [11] with a possible phenotypic effect.
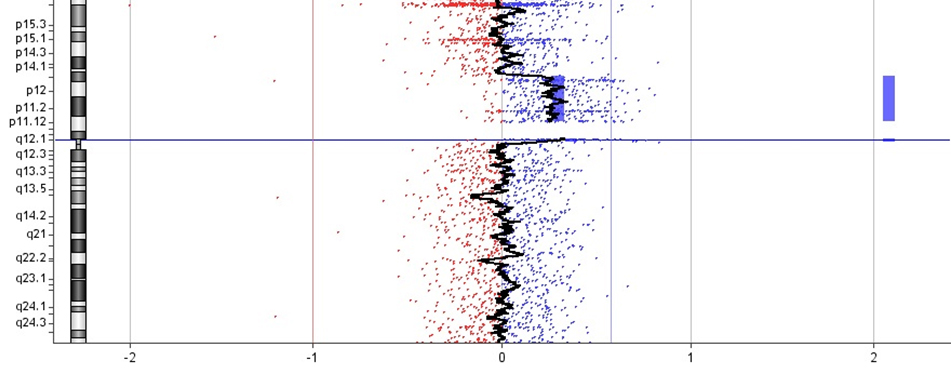
Figure 2 Graph of the array-CGH of chromosome 11.
After genetic counseling, the parents decided to terminate the pregnancy at 18 w.o.g. A small tissue sample was used for culture preparation. Again a karyotype of 47,XY, +mar (dn)/46,XY was found. Autopsy showed a female fetus, too small for its gestational age, with oval face, low-set ears and long philtrum. No other pathological findings were detected in the other organs. The histological examination of the placenta showed immature and dysmorphic villi.
4. Discussion
sSMCs are a major clinical problem in prenatal genetic diagnosis and counseling. Their detailed characterization is required to establish reliable genotype-phenotype correlations in future. At present it is known that in de novo sSMCs, the overall risk for an abnormal phenotype ranges between 14% and 30% depending on the genetic content of the sSMC [5,8,9,10]. Moreover, if high resolution ultrasound studies are normal, this risk decreases to 18% [9]. The models of how de novo sSMC could be formed are based in part on the coexistence of uniparental disomy and sSMC, and on the observation that sSMC could be evolved due to incomplete trisomic rescue. One or combination of these models happen as a result of the de novo sSMC formation during gametogenesis or embryogenesis [11]. It is important to note that conventional cytogenetic techniques are not sufficient to characterize almost all sSMCs.
In recent years the utility of chromosomal microarray analysis and FISH techniques have been found helpful in correlating the genotype with the phenotype of a patient [12,13]. Genotype-phenotype correlation of sSMCs is partially hindered e.g. by the influence of mosaicism in different tissues and the complexity of sSMC uniparental disomy [14,15].
According to the literature [16], there are only 40 sSMC cases derived from chromosome 11 (sSMC [11]) reported yet. Eight sSMC [11] were without and 10 cases with clinical findings, while in 20 cases there was no or unclear clinical correlation. Also, there are 2 cases with neocentromeres. Most of the cases with clinical findings include hypotonia, dysmorphic face, developmental delay and brain malformations [16]. At the present case the fetus was small, possibly due to in utero growth retardation, and it had dysmorphic facial features, as the pregnancy was terminated at 18 w.o.g. its contribution for genotype-phenotype correlation may be only minor. Still it is obvious that partial trisomy including chr11:30,796,545-56,649,983 leads to clinical adverse effects. However, according to [16] the region 11q12.1, being trisomic due to sSMC presence is not leading to any clinical problems. Thus, only cases with interstitial duplication of 11p14 to 11p11.2 and clinical signs can be compared to the present case. Palutke et al. (1980) reported a female patient with a dup [11](p12p14) with abnormal eye movements, ventricular septal defect and dysmorphic features [17]. Also there are 4 cases with dup [11](p11.3p11.3) with variant clinical features, like macular dysfunction, cleft lip and palate and developmental delay [18], mentally retardation but no other remarkable dysmorphic characteristics [19,20], or features invoking Silver-Russell syndrome [21].
The genes being duplicated due to sSMC presence in this case might have had severe effects at the postnatal life. However, only three genes in the duplicated region were specifically correlated with abnormalities when being upregulated. These are ALX4, CELF1 and Pax6. The first is correlated with craniosynostosis, a condition describing the early fusion of one or more sutures of the infant skull [22], while the second is linked with myotonic dystrophy type 1 [23,24], a muscle disease characterized by various degrees of muscle weakness, arrhythmia and/or cardiac conduction disorders, cataract, endocrine damage, sleep disorders and baldness. Pax6, a transcription factor which plays role in the eye [reviewed in [25]], pancreas [reviewed in [26]], olfactory system [reviewed in [27]] and cortex development [reviewed in [28]] was found to be duplicated in a girl with seizures, developmental delay, microcephaly and minor ocular findings, while her optical nerve, macula, and retinal vessels were normal [29]. Another study correlated an 11p13-12 tandem duplication found in a girl with borderline developmental delay, mild facial anomalies and eye abnormalities including the Pax6 gene [30].
According to autopsy, the examined fetus appeared to be small for its gestational age. The genes CREB3L1, LRP4, SLC39A13 contribute to normal growth, as homozygous deficient mice for these genes exhibited growth retardation and bone deformities [31,32,33]. Despite these findings, it remains to be elucidated if the genes in the duplicated region have the same dosage effects if they are present trisomic due to a mosaic sSMC, a tandem duplication, or a duplication inserted elsewhere, having position effects. Although, the fetus, with the largest sSMC [11] is reported here, had a mild phenotype, we propose that the mild phenotype might be attributed to the mosaicism of the sSMC.
5. Conclusions
As the minute sSMC [11] reported here was present only in 10-15% mosaic state, aCGH could also easily have failed to characterize its origin [34]. Thus, molecular cytogenetics and molecular karyotyping are powerful tools for sSMC characterization. Both can contribute to genotype-phenotype correlations. Overall, application of a straightforward diagnostic scheme to characterize an sSMC’s chromosomal origin and content to be able to compare with data from the literature [35] is imperative, especially in prenatal sSMC cases.
Acknowledgments
The authors would like to thank the family with the sSMC [11] fetus for their collaboration.
Author Contributions
AP, KT, TS carried out the chorionic villi sampling and the amnioncentesis. TD and GP examined the fetus. ΜΚ, VP and AA wrote the manuscript. ES and IP carried out the molecular karyotype analysis. AZ and LT read the manuscript critically. EM carried out the conventional karyotype analysis and conducted the study.
Funding
Access to Genome – ATG P.C. Athens – Thessaloniki, Greece.
Competing Interests
The authors have declared that no competing interests exist.
References
- Liehr T, Claussen U, Starke H. Small supernumerary marker chromosomes (sSMC) in humans. Cytogenet Genome Res. 2004; 107: 55-67. [CrossRef]
- Liehr T, Weise A. Frequency of small supernumerary marker chromosomes in prenatal, newborn, developmentally retarded and infertility diagnostics. Int J Mol Med. 2007; 19: 719-731. [CrossRef]
- Pietrzak J, Mrasek K, Obersztyn E, Stankiewicz P, Kosyakova N, Weise A, et al. Molecular cytogenetic characterization of eight small supernumerary marker chromosomes originating from chromosomes 2, 4, 8, 18, and 21 in three patients. J Appl Genet. 2007; 48: 167-175. [CrossRef]
- Blennow E, Bui TH, Kristoffersson U, Vujic M, Annerén G, Holmberg E, et al. Swedish survey on extra structurally abnormal chromosomes in 39 105 consecutive prenatal diagnoses: prevalence and characterization by fluorescence in situ hybridization. Prenat Diagn. 1994; 14: 1019-1028. [CrossRef]
- Crolla JA. FISH and molecular studies of autosomal supernumerary marker chromosomes excluding those derived from chromosome 15: II. Review of the literature. Am J Med Genet. 1998; 75: 367-381. [CrossRef]
- Manolakos E, Kefalas K, Vetro A, Oikonomidou E, Daskalakis G, Psara N, et al. Prenatal diagnosis of two de novo 4q35-qter deletions characterized by array-CGH. Mol Cytogenet. 2013; 6: 47. [CrossRef]
- McGowan-Jordan J, Simons A, Schmid M. An International System for Human Cytogenomic Nomenclature (2016). Karger Publishing
- Crolla JA, Youings SA, Ennis S, Jacobs PA. Supernumerary marker chromosomes in man: parental origin, mosaicism and maternal age revisited. Eur J Hum Genet. 2005; 13: 154-160. [CrossRef]
- Graf MD, Christ L, Mascarello JT, Mowrey P, Pettenati M, Stetten G, et al. Redefining the risks of prenatally ascertained supernumerary marker chromosomes: a collaborative study. Med Genet. 2006; 43: 660-664. [CrossRef]
- Warburton D. De novo balanced chromosome rearrangements and extra marker chromosomes identified at prenatal diagnosis: clinical significance and distribution of breakpoints. Am J Hum Genet. 1991; 49: 995-1013.
- Liehr T. Small supernumerary marker chromosomes (sSMC) a guide for human geneticists and clinicians; with contributions by UNIQUE (The Rare Chromosome Disorder Support Group). Springer, 2012, Chapter 3, p.21.
- Marle N, Martinet D, Aboura A, Joly-Helas G, Andrieux J, Flori E, et al. Molecular characterization of 39 de novo sSMC: contribution to prognosis and genetic counselling, a prospective study. Clin Genet. 2014; 85: 233-244 [CrossRef]
- Vetro A, Manolakos E, Petersen MB, Thomaidis L, Liehr T, Croci G, et al. Unexpected results in the constitution of small supernumerary marker chromosomes. Eur J Med Genet. 2012; 55: 185-190. [CrossRef]
- Tsuchiya KD, Opheim KE, Hannibal MC, Hing AV, Glass IA, Raff ML, et al. Unexpected structural complexity of supernumerary marker chromosomes characterized by microarray comparative genomic hybridization. Mol Cytogenet. 2008; 21: 1-7. [CrossRef]
- Yu S, Fiedler SD, Brawner SJ, Joyce JM, Zhou XG, Liu HY. Characterizing small supernumerary marker chromosomes with combination of multiple techniques. Cytogenet Genome Res. 2012; 136: 6-14. [CrossRef]
- Liehr T. 2018. Small supernumerary marker chromosomes. http://ssmc-tl.com/sSMC.html small supernumerary marker chromosomes. Accessed: 10/02/2018)
- Palutke W, Tyrkus M, Gohle N, Bawle E, Woolley PV. Intra-segmental duplication resulting in partial trisomy 11p: case report and cytogenetic documentation. Am J Hum Genet. 1980; 32: 83A.
- Strobel RJ, Riccardi VM, Ledbetter DH, Hittner HM. Duplication 11p11.3 leads to 14.1 to meiotic crossing--over. Am J Med Genet. 1980; 7: 15-20. [CrossRef]
- Lavedan C, Barichard F, Azoulay M, Couillin P, Molina Gomez D, Nicolas H, et al. Molecular definition of de novo and genetically transmitted WAGR-associated rearrangements of 11p13. Cytogenet Cell Genet. 1989; 50: 70-74. [CrossRef]
- Speleman F, Mannens M, Redeker B, Vercruyssen M, Van Oostveldt P, Leroy J, et al. Cytogenet characterization of a de novo duplication of 11p14----p13, using fluorescent in situ hybridization and southern hybridization. Cell Genet. 1991; 56: 129-131. [CrossRef]
- Palumbo O,Mattina T,Palumbo P, Carella M, Perrotta CS. A de novo 11p13 Microduplication in a patient with some features invoking silver-russell syndrome. Mol Syndromol. 2014; 5: 11-18.
- Yagnik G, Ghuman A, Kim S, Stevens CG, Kimonis V, Stoler J, Sanchez-Lara PA, et al. ALX4 gain-of-function mutations in nonsyndromic craniosynostosis. Hum Mutat. 2012; 33: 1626-1629 [CrossRef]
- Koshelev M, Sarma S, Price RE, Wehrens XH, Cooper TA. Heart-specific overexpression of CUGBP1 reproduces functional and molecular abnormalities of myotonic dystrophy type 1. Hum Mol Genet. 2010; 19: 1066-1075. [CrossRef]
- Ward AJ, Rimer M, Killian JM, Dowling JJ, Cooper TA. CUGBP1 overexpression in mouse skeletal muscle reproduces features of myotonic dystrophy type 1. Hum Mol Genet. 2010; 19: 3614-3622. [CrossRef]
- Cvekl A, Callaerts P PAX6: 25th anniversary and more to learn. Exp Eye Res. 2017; 156: 10-21. [CrossRef]
- Dohrmann C, Gruss P, Lemaire L. Pax genes and the differentiation of hormone-producing endocrine cells in the pancreas. Mech Dev. 2000; 92: 47-54. [CrossRef]
- Nomura T, Haba H, Osumi N. Role of a transcription factor Pax6 in the developing vertebrate olfactory system. Dev Growth Differ. 2007; 49: 683-690. [CrossRef]
- Manuel MN, Mi D, Mason JO, Price DJ. Regulation of cerebral cortical neurogenesis by the Pax6 transcription factor. Front Cell Neurosci. 2015; 9: 70. [CrossRef]
- Aradhya S, Smaoui N, Marble M, Lacassie Y. De novo duplication 11p13 involving the PAX6 gene in a patient with neonatal seizures, hypotonia, microcephaly, developmental disability and minor ocular manifestations. Am J Med Genet A. 2011; 155A: 442-444. [CrossRef]
- Aalfs CM, Fantes JA, Wenniger-Prick LJ, Sluijter S, Hennekam RC, van Heyningen V, et al. Tandem duplication of 11p12-p13 in a child with borderline development delay and eye abnormalities: dose effect of the PAX6 gene product? Am J Med Genet. 1997; 73: 267-271. [CrossRef]
- Fukada T, Civic N, Furuichi T, Shimoda S, Mishima K, Higashiyama H, et al. The zinc transporter SLC39A13/ZIP13 is required for connective tissue development; its involvement in BMP/TGF-beta signaling pathways. Plos One. 2008; 3: e3642. [CrossRef]
- Johnson EB, Hammer RE, Herz J. Abnormal development of the apical ectodermal ridge and polysyndactyly in Megf7-deficient mice. Hum Mol Genet. 2005; 14: 3523-3538. [CrossRef]
- Murakami T, Saito A, Hino S, Kondo S, Kanemoto S, Chihara K, Sekiya H, et al. Signalling mediated by the endoplasmic reticulum stress transducer OASIS is involved in bone formation. Nat Cell Biol. 2009; 11: 1205-1211. [CrossRef]
- Reddy KS, Aradhya S, Meck J, Tiller G, Abboy S, Bass H. A systematic analysis of small supernumerary marker chromosomes using array CGH exposes unexpected complexity. Genet Med. 2013; 15: 3-13. [CrossRef]
- Liehr T. Characterization of prenatally assessed de novo small supernumerary marker chromosomes by molecular cytogenetics. Methods Mol Biol. 2008; 444: 27-38. [CrossRef]

