

Reminiscence from Half a Century of Anti-Pneumocystis Drug Discovery and Development
Walter Hughes * ![]()
- Department of Infectious Diseases, St. Jude Children’s Research Hospital, Memphis, Tennessee, USA
* Correspondence: Walter Hughes ![]()
Received: September 12, 2018 | Accepted: November 30, 2018 | Published: December 05, 2018
OBM Genetics 2018, Volume 2, Issue 4 doi: 10.21926/obm.genet.1804052
Academic Editors: Andrés Moya, Enrique J. Calderón and Luis Delaye
Special Issue: Pneumocystis: A Model of Adaptive Coevolution
Recommended citation: Hughes W. Reminiscence from Half a Century of Anti-Pneumocystis Drug Discovery and Development. OBM Genetics 2018;2(4):052; doi:10.21926/obm.genet.1804052.
© 2018 by the authors. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
Abstract
My experiences with the identification and development of the anti-Pneumocystis drugs trimethoprim-sulfamethoxazole, dapsone and atovaquone are recounted. Attention is drawn to certain often-overlooked aspects of these drugs, including their concomitant effects against infections other than Pneumocystis, matters of dosing and adverse effects. During the past four decades several million people worldwide received chemoprophylaxis and treatment with these drugs for Pneumocystis jirovecii pneumonia. The population of immunocompromised patients susceptible to this infection is expected to increase in the future and new drugs may be needed. Despite the discovery of many promising new anti-Pneumocystis drugs in the laboratory, none has received FDA approval for clinical use within the past 25 years. Opportunities for translational studies to advance these discoveries and others to clinical application are discussed.
Keywords
Pneumocystis jirovecii; pneumocystis carinii; pneumonia; trimethoprim-sulfamethoxazole; atovaquone; dapsone; pentamidine
1. Introduction
During the past one-half century, the prevalence of Pneumocystis jirovecii pneumonia (PCP) has progressively increased from a few hundred to a few million cases. Populations at risk have included debilitated infants in European nurseries, children with congenital immunodeficiency disorders, patients with cancer, organ transplant recipients and those receiving immunosuppressive drugs for a variety of diseases. No group has been more vulnerable and numerous than patients with the acquired immunodeficiency syndrome (AIDS). Recent advances in the treatment of several chronic diseases cause suppression of the immune system and add new categories of individuals at risk for PCP. Rheumatologists are now pondering over guidelines for the use of PCP chemoprophylaxis for patients with rheumatoid arthritis, one of the most common chronic diseases of humans. Considering the fact that at least 75 % of normal children worldwide have subclinical infection with P. jirovecii by four years of age and possibly retain latent organisms for life, plus the expectations for a forthcoming plethora of new therapeutic modalities that compromise host defence, it seems likely the prevalence of PCP will continue to increase.
No new drug for the treatment or prevention of PCP has been approved by the U S Food and Drug Administration (FDA) during the last 25 years, the last being atovaquone (Mepron™) and trimetrexate (Neu Trexin™) in 1993. Despite the discovery of several dozen new anti-PCP compounds in the laboratory during the past quarter century, none has achieved FDA certification for use in clinical practice. Approval by the FDA of a drug for a specific indication is required before physicians are willing or allowed to prescribe it. The use of drugs not approved by the FDA carries medico-legal liabilities. Stringent clinical research trials are required for FDA certification.
Although the ultimate objective of basic and clinical research with P. carinii/jirovecii is to benefit the health and welfare of humans, translational studies to bridge the laboratory and clinic have been woefully lacking regarding treatment and prevention. There are good reasons for this lag. This commentary aims to review aspects of the early development of three drugs now in use for the treatment and prevention of PCP and to address the formidable problems to be faced today in new clinical trials. The commentary is not intended to be a comprehensive review of all studies in drug development for this disease.
For more than 40 years trimethoprim-sulfamethoxazole (TMP-SMZ) has been the unchallenged drug of first choice for the treatment and prophylaxis of PCP. During this time, it has been administered to millions of people worldwide, often for long courses spanning months and years. The drug is highly effective, relatively safe, readily available, inexpensive and can be administered orally or intravenously. The formulation, doses and intervals of administration used today are essentially those used in the original studies [1,2,3,4,5,6,7]. Furthermore, two of the drugs currently recommended as alternatives to TMP-SMZ, namely dapsone and atovaquone, have been in use for some 30 and 25 years, respectively [8,9,10,11,12,13]. Pentamidine isethionate, the first drug used to treat PCP in 1958, continues in use after 60 years [14]. It is recommended as an alternative to TMP-SMZ but is limited in use due to severe toxicity from the parenteral route and the need for aerosol administration as prophylaxis. These drugs are recommended in guidelines from the Centres for Disease Control and Prevention (CDC), the National Institutes of Health (NIH), the Infectious Diseases Society of America [15] and the World Health Organization (WHO) for the treatment and prevention of PCP in AIDS. Trimetrexate is not recommended because of limited efficacy, adverse effects and termination of production by the manufacturer. The history of early circumstances, discovery and development of TMP-SMZ, dapsone and atovaquone will be reviewed with attention to certain aspects often overlooked.
2. Trimethoprim-Sulfamethoxazole
In 1962 the new St. Jude Children’s Research Hospital opened its doors for research in catastrophic diseases of children, primarily cancer. Success came quickly with the discovery and development of the “Total Therapy” regimen to cure acute lymphocytic leukaemia, a heretofore universally fatal disease. Worldwide referral of children with leukaemia led to the accumulation of a relatively large number of heavily immunosuppressed patients at this centre to receive the new therapy. In 1967 a case of PCP occurred and two more in 1968. In 1970 the CDC reported in the Journal of the American Medical Association that “the first large outbreak (19 cases) of Pneumocystis carinii pneumonia reported in the United States occurred at St. Jude Children’s Hospital in Memphis” [16]. By 1974 forty cases of PCP were occurring per year in the rapidly growing population of immunosuppressed children.
The pneumonitis was fatal in 100 % of cases, if untreated. It was unique among other opportunistic infections in that the organism and the disease it caused remained localized to the lungs, even in fatal cases. Furthermore, it occurred in cancer patients during remission as well as relapse while on chemotherapy [17].
At the time, the only drug known to have some therapeutic effect on PCP was pentamidine isethionate (Lomidine™), a parenteral anti-protozoan diamidine from May & Baker, Ltd. in England. Because of its severe toxicity it was not approved by the FDA for use in the United States, but was made available as an intramuscular injection on a case-by-case basis through the CDC in Atlanta. PCP was occurring in about 20 % of children with acute lymphocytic leukaemia and thwarted successful chemotherapy now curative for the malignancy. St. Jude investigators began research efforts to find a means to effectively prevent and treat PCP.
The first breakthrough came when the investigators learned of two obscure European reports by Weller in 1955-56 [18,19], wherein rats being administered cortisone acetate acquired PCP spontaneously without inoculation. Furthermore, Frenkel et al, in 1966 treated such cortisone-immunosuppressed rats with several drugs and reported that 6 rats given pyrimethamine and sulfadiazine survived without PCP while 6 untreated controls died of PCP [20]. This led the St. Jude group to begin the use of animal experimentation in search of a solution to their problem with PCP. While P. carinii infection in the rat histopathologically resembled that in man, no corollary clinical studies had been done at that time to establish it as a valid animal model for human PCP.
St. Jude investigators initially evaluated a vaccine prepared from whole cell P. carinii organisms harvested by lavage from rat lungs. Normal immunocompetent rats were immunized to achieve a protective antibody level, then immunosuppressed with cortisone acetate and given tetracycline to prevent bacterial infections for 6 weeks, at which time all had become infected with extensive PCP [21]. While immunization was ineffective in preventing PCP, an interesting incidental observation was made from the studies, as reported by Kim, et al in 1972 [22]. They studied 8 rat and 5 human isolates of Pneumocystis sp. with antisera raised to rat organisms, and with indirect fluorescent antibody methods found that “all of the rat strains fluoresced while none of human strains gave fluorescence, which suggested antigenic dissimilarity between these two strains.” This was the first sound evidence for a difference in rat and human strains, later substantiated by genetic analysis leading to a change in nomenclature with P. carinii to designate the rat strain and P. jirovecii the human strain [23].
The investigators abandoned the vaccine effort and directed their attention to a search for a new and more effective drug to treat and prevent PCP. They reasoned that if a drug to be tested were administered to rats treated with cortisone for 6 weeks and at autopsy was found to have no PCP, in contrast to PCP in 90-100 % of untreated controls, one could conclude the test drug had anti-P. carinii activity. After a relentless search of many drugs and compounds selected by deductive reasoning that drugs effective against protozoal infections such as malaria, trypanosomiasis, etc. might be effective against the presumed protozoan Pneumocystis carinii, the drug combination of trimethoprim-sulfamethoxazole (TMP-SMZ) was found to be 100 % effective in this and repeated experiments (Table 1) [3].
Table 1 Effects of trimethoprim-sulfamethoxazole in prevention and treatment of P. carinii infection in rats [3].
|
Group |
Percent rats with P. carinii Pneumonitis |
|
Control (no cortisone) |
0 % |
|
Control (cortisone x 6 wk. ) |
100 % |
|
Trimethoprim-sulfamethoxazole prophylaxis + cortisone |
0 % |
|
Trimethoprim-sulfamethoxazole treatment* + cortisone |
36% |
|
Pentamidine prophylaxis + cortisone |
86 % |
*TMP-SMZ delayed until first animal in group died with PCP.
Further studies showed trimethoprim (TMP) alone to be totally ineffective, but served synergistically to enhance the activity of sulfamethoxazole (SMZ). This antibiotic (Cotrimoxazole) was beginning to be used in Europe for the treatment of bacterial infections of the urinary tract in adults, but had not reached FDA approval for that use in the United States. It was chosen for the PCP rat study because a few anecdotal reports of its success in treating malaria. The FDA readily approved Investigational New Drug (IND) status for inhouse clinical trials at St. Jude to evaluate TMP-SMZ for the treatment and prevention of PCP.
The first study, a proof of principal design, showed TMP-SMZ to be effective in the treatment of 20 patients with PCP, based on the knowledge that untreated the infection was 100 % fatal (Table 2) [4].
Table 2 First trial of TMP-SMZ for the treatment PCP in humans; open label, proof of principal [4].
|
Group |
No. Patients with PCP |
Dose TMP-SMZ mg/kg/day |
No. (%) Recovered |
|
High Dosage |
14 |
20 mg TMP-100 mg SMZ |
12 (86 %) |
|
Low Dosage |
6 |
4-7 mg TMP-20-35 mg SMZ |
4 (67 %) |
|
Total |
20 |
4-20 mg TMP-20-100 mg SMZ |
16 (80 %) |
The second study compared TMP-SMZ with pentamidine isethionate in the treatment of PCP in children with cancer. Fifty patients were randomized to receive 20 mg TMP-100 mg SMZ per kg per day or pentamidine isethionate 4.0 mg per kg per day for 14 days. The results are summarized in Table 3 [5].
Table 3 Comparison of TMP-SMZ with pentamidine isethionate in the treatment of 50 patients with PCP [5].
|
Treatment Group |
No. Patients |
No. (%) Recovered from PCP |
No. (%) Adverse effects |
|
TMP-SMZ |
26 |
20 (77 %) |
1/17 (6 %) |
|
Pentamidine |
24 |
18 (75 %) |
14/15 (93 %) |
A pivotal study was then designed to determine the efficacy and feasibility of the long-term (at least two years) administration of TMP-SMZ to prevent PCP in children with cancer.
Between 1974 and 1976 a randomized, double-blind, placebo-controlled study evaluated the efficacy of TMP-SMZ for the prevention of PCP in 160 cancer patients at high risk (>15 %) for PCP at St. Jude. Eighty patients were randomized to receive 150 mg TMP and 750 mg SMZ per square meter body surface per day (equal to 20 mg TMP- 100 mg SMZ / kg/day) orally and 80 additional patients received a placebo for two years [1]. Note the method used for dose calculation, to be discussed later.
The placebo-controlled design of this study, not possible with subsequent human studies, once efficacy had been demonstrated, provided perhaps the most precise data available on the adverse reactions and safety of TMP-SMZ. These data are as valid and pertinent today as when they were derived. It is important to clinical practice today and to future clinical trials to mention how patients were prospectively evaluated for adverse effects, because many of the subsequent clinical trials have been flawed in assessing this aspect. At each clinic visit patients were asked about specific adverse effects, including rash, jaundice, diarrhoea, vomiting, abdominal pain, headache, lethargy, nausea and oral lesions or glossitis. Complete blood counts were done weekly; and, chest radiographs, bone-marrow examinations, renal and liver function tests, serum folic acid, total protein, albumin, globulin, immunoglobulins G, A and M, and cultures for bacteria and fungi of the pharynx and rectum were taken at three-month intervals. The diagnosis of PCP required the demonstration of pneumonitis by chest radiograph and Pneumocystis organisms from lung specimens (biopsy, needle aspirate, bronchoalveolar lavage).
At the end of the first year of the study, planned for two years, statisticians concluded that from the overall results to date efficacy could be established, that the code could be broken, and the study could be terminated. The investigators argued to continue the study for another year. They feared some significant adverse events might occur with long-term administration that could offset the benefit of preventing PCP. Adverse events were being recorded in 15 to 20 % of cases. Of special concern were possible changes in microbial flora due to the broad-spectrum antibiotic effect of TMP-SMZ, emergence of drug-resistant bacteria or Pneumocystis sp., fungal overgrowth with serious mycotic infections, and other adverse effects that might occur during the period of two and one-half years or longer needed to cover the course of anticancer chemotherapy. The ethical issue was that continuing the study another year would deprive patients at risk for PCP of protection. On the other hand, once TMP-SMZ was shown to have some protective efficacy, no future studies with placebo comparison would be ethical. The investigators prevailed and continued the study for another year. The code was broken in October 1976.
Results revealed that PCP developed in 17 (21 %) of the 80 patients in the placebo group, in contrast with none of the 80 patients in the group treated with TMP-SMZ (P<0.01). TMP-SMZ was 100 % effective in the prevention of PCP (Table 4).
Table 4 Results of a randomized, double-blind, placebo-controlled study at St. Jude Children’s Research Hospital of 160 cancer patients randomized to receive placebo or TMP-SMZ prophylactically to prevent PCP [1].
|
Group |
No. Patients |
Days on Study |
No. (%) with PCP* |
|
Placebo |
80 |
27,138 |
17 (21%) |
|
Trimethoprim-sulfamethoxazole |
80 |
30,589 |
0 (0%) |
*P< 0.01
An important aspect of the prophylaxis study, often overlooked today, is the broad-spectrum antimicrobial effects of TMP-SMZ, both bacterial and protozoan. TMP-SMZ is known to be effective against Streptococcus pneumoniae, Haemophilus influenzae, Escherichia coli, Klebsiella sp, Enterobacter, sp, Morganella sp, Proteus sp, and Shigella sp. It is now approved by the FDA for the treatment of urinary tract infections, acute otitis media, bronchitis, traveller’s diarrhoea due to enterotoxigenic E. coli and Shigellosis, as well as treatment and prevention of PCP. It has some efficacy in the treatment of other bacterial infections, toxoplasmosis and malaria. This placebo-controlled study showed the impact of TMP-SMZ prophylaxis on certain other infections common to the immunocompromised host (Tables 5-6).
Table 5 Types of non-PCP infections in patients treated with placebo and trimethoprim-sulfamethoxazole (TMP-SMZ) [1].
|
Category |
Episodes per 30,000 Patient Days |
|
|
Placebo |
TMP-SMZ |
|
|
Upper Respiratory Tract Infection |
111 |
50* |
|
Acute Otitis Media |
50 |
6* |
|
Pneumonia (Not PCP) |
21 |
8* |
|
Bronchitis |
17 |
11 |
|
Laryngotracheobronchitis |
1 |
1 |
|
Conjunctivitis |
3 |
0 |
|
Dental Abscess |
3 |
0 |
|
Enteritis |
19 |
14 |
|
Sinusitis |
16 |
8* |
|
Hepatitis |
3 |
0 |
|
Pancreatitis |
2 |
0 |
|
Sub-hepatic Abscess |
0 |
1 |
|
Cellulitis |
25 |
9* |
|
Urinary Tract Infections |
2 |
5 |
|
Osteomyelitis |
0 |
1 |
|
Aseptic Meningitis |
1 |
2 |
|
Fever of Undetermined Origin |
69 |
61 |
*P< 0.01
Bacterial sepsis, pneumonia other than PCP, acute otitis media, upper respiratory tract infection, sinusitis, and cellulitis occurred significantly less often in TMP-SMZ-treated than the placebo groups. Oral candidiasis occurred more frequently in the drug-treated group. These factors must be kept in mind when selecting PCP prophylaxis for immunocompromised patients.
Monitoring the pharyngeal and rectal microbial flora during the two-year study showed no significant difference in colonization rates at any time for Pseudomonas aeruginosa, C. albicans, Candida species other than albicans, alpha-haemolytic streptococcus, Staphylococcus epidermidis, Streptococcus pneumoniae, and aspergillus sp. Importantly, there was a significant reduction in pharyngeal colonization with Staphylococcus aureus and rectal colonization with Escherichia coli, Proteus mirabilis, and Klebsiella pneumoniae. In sum, there was no significant increase for any organism in the group treated with TMP-SMZ.
Table 6 Infections with specific causative agents in placebo and trimethoprim-sulfamethoxazole (TMP-SMZ) groups [1].
|
Category |
Episodes per 30,000 Patient Days |
|
|
Placebo |
TMP-SMZ |
|
|
Bacterial sepsis (total) |
16 |
6* |
|
Pseudomonas aeruginosa |
3 |
1 |
|
Klebsiella pneumoniae |
2 |
1 |
|
Streptococcus pneumoniae |
2 |
0 |
|
Escherichia coli |
3 |
0 |
|
Staphylococcus aureus |
3 |
1 |
|
Streptococcus, Group B |
0 |
1 |
|
Mixed |
2 |
1 |
|
Gram-negative bacillus |
1 |
1 |
|
Streptococcus beta haemolytic (Group A) tonsillitis |
4 |
0 |
|
Shigella enteritis |
1 |
0 |
|
Varicella |
4 |
4 |
|
Zoster |
11 |
14 |
|
Herpes labialis |
20 |
8 |
|
Herpetic gingivostomatitis |
3 |
1 |
|
Herpangina |
1 |
0 |
|
Mumps |
1 |
0 |
|
Oral candidiasis |
13 |
31* |
|
Pulmonary candidiasis |
1 |
2 |
|
Oesophageal candidiasis |
1 |
1 |
|
Tinea corporis |
3 |
1 |
|
Disseminated mycoses (total) |
3 |
4 |
|
C. albicans |
0 |
3 |
|
C. tropicalis |
1 |
0 |
|
C. guilliermondii & aspergillus species |
0 |
1 |
|
Cryptococcus neoformans |
1 |
0 |
|
Histoplasma capsulatum |
1 |
0 |
*P< 0.02
Adverse effects of rash, vomiting, diarrhoea, abdominal pain, nausea, headache, oral lesions, glossitis and jaundice; and, abnormalities of haematological, folic acid, renal and hepatic profiles occurred no more frequently in the TMP-SMZ group than in the placebo group. Two patients were withdrawn from the study for possible life-threatening adverse events: one with recurrent urticarial rash and another with prolonged neutropenia. Both patients had received the placebo.
Upon completion of the prophylaxis trial all cancer patients at St. Jude who were at high risk for PCP (> 15 %) were given TMP-SMZ daily while on immunosuppressive therapy. The impact is shown in Figure 1. TMP-SMZ brought about an abrupt cessation of the epidemic, despite a progressive increase in number of cancer patients at risk and intensity of immunosuppressive chemotherapy. The investigators reported a period of five years without a single case of PCP [24,25]. The TMP-SMZ prophylaxis regimen was studied at other cancer centres [26,27,28] with results like those at St. Jude. Thereafter, PCP prophylaxis became a part of supportive management as indicated for cancer chemotherapy as well as other conditions subject to PCP.

Figure 1 Panoramic graph of incidence and prevalence of PCP at St. Jude Children’s Research Hospital from 1967 (first case) through 1985 [1,29]. The TMP-SMZ/placebo study was from October 1974 through October 1976, after which all patients at high risk for PCP were given TMP-SMZ prophylaxis. White dots indicate the expected number of cases of PCP based on incidence before onset of study.
After a couple of years or so some concern arose in the minds of the St. Jude investigators. Something just didn’t seem right. How could there be no cases of PCP? No drug is perfect and compliance with administration is always fraught with gaps of illness and negligence. Could the widespread use of TMP-SMZ have eradicated Pneumocystis carinii from the environment and it was no longer a factor? Another possibility was that maybe the dose and/or frequency of TMP-SMZ was far more than what was needed; so, if periods of non-compliance, or missed doses occurred, the protective effect would not be lost. They then resorted back to the rat model.
When cortisone-treated rats were given TMP-SMZ only three days a week and none for the remaining four, the results were the same as for daily doses, providing effective prophylaxis for PCP in 100 % of animals [30]. Because their earlier studies had by now validated the cortisone-treated rat as an animal model for the human disease, enough confidence had accrued for the St. Jude investigators to proceed with a clinical trial comparing daily and intermittent three-days-a-week regimens.
In a prospective, randomized, two-year clinical trial in children with acute lymphocytic leukaemia no cases of PCP occurred in any of the 92 patients receiving TMP-SMZ daily or in any of the 74 who received it three consecutive days per week; whereas, the expected incidence of PCP with no prophylaxis was 21 % [7]. Importantly, 10 cases of systemic mycoses occurred in the daily group and only one in the three-days-a-week group (P = 0.024). There were no differences in the rates of other infections or adverse effects associated with the drug. It was concluded that the intermittent schedule had the advantage of less frequent fungal infections and lower cost but did not likely allow for as great a buffer for missed doses of TMP-SMZ.
The remarkable correlation of the experiments in the cortisone-treated rat and clinical trials with identical experimental design and results is to be noted. These studies clearly established the cortisone-treated rat as an animal model for human Pneumocystis pneumonia. Confidence in the rat model was important to future development of anti-Pneumocystis therapy to be described.
For a brief time during the latter part of the 1970s, the problem of PCP seemed to have diminished to a reasonably controllable infectious disease. However, in 1980 a new era began for PCP thrusting it to major prominence worldwide. Doctors in California and New York began to encounter cases of PCP in otherwise healthy men. They did not have cancer, congenital immunodeficiency disorders, were not organ transplant recipients and were not receiving immunosuppressive drugs. Knowing PCP did not occur in otherwise normal individuals, the astute doctors searched for an underlying condition, leading to the discovery of the new acquired immunodeficiency syndrome (AIDS) [31,32]. In a sense, PCP discovered AIDS, because the human immunodeficiency virus (HIV) infection is silent with no symptoms or discernible disease even at autopsy.
Of the first 1000 cases of AIDS, 50 % had PCP [33]. Two aspects of PCP cases with AIDS differed somewhat from PCP in the past, and possibly one was related to the other. Firstly, prior to the AIDS epidemic PCP occurred predominantly in infants and children, namely nursery outbreaks in Europe, children with congenital immunodeficiency disorders, cancer and severe malnutrition. PCP with AIDS occurred almost exclusively in adults. Secondly, higher rates of adverse reactions to TMP-SMZ were encountered in the HIV-infected individuals [33,34]. For example, the report of Jaffee, et al [33] described adverse reactions of fever, malaise, nausea, headaches, rash and cytopenias in 8 (44 %) of 18 patients with PCP and AIDS treated with 20 mg/kg TMP- 100 mg/kg SMZ per day, intravenously. This article and others were influential in causing physicians to limit the use of TMP-SMZ in AIDS patients, and some resorted to pentamidine, still only available from the CDC.
It is sufficiently noteworthy to stop here and comment on one of the remarkable circumstances associated with the use of TMP-SMZ in the early days of the AIDS epidemic, and possibly to some extent even today. It stems from the fact that the original clinical trials on the use of TMP-SMZ for PCP at St. Jude were used by the FDA to approve TMP-SMZ for treatment of PCP. In design, two factors from these studies were of later influence. Firstly, the St. Jude investigators had used the maximal tolerated dose of TMP-SMZ for their studies to avoid the possibility of inadequate dosing being a factor if efficacy was not demonstrated. Secondly, all dosing in paediatrics is based on body weight or body surface area. This calculation for doses of drugs is applied until one fixed dose, used for all adults, is reached. The paediatric dosages by body weight cannot be applied to adults. For comparison, the previous FDA approved standard adult dose for TMP-SMZ was 320 mg TMP- 1600 mg SMZ total dose per day in the treatment of bacterial infections and the paediatric dose was 8.0 mg/kg TMP – 40.0 mg/kg SMZ per day for this purpose. Keep in mind the PCP investigators had arbitrarily used the high maximally tolerated dose of 20 mg/kg TMP-100 mg/kg SMZ per day for their clinical trials to determine the efficacy for PCP. This was two and one-half times greater than the standard dose used to treat bacterial infections in children. The story continues.
The FDA regulations require the dosage used in the determinative research trials be used in the final FDA label. Thus, the dosage approved by the FDA for TMP-SMZ treatment of PCP was 20 mg/kg TMP – 100 mg/kg SMZ per day (equal to 150 mg/ M2 TMP – 750 mg/ M2 SMZ). This dosing was unusual because almost all the drug applications are based on studies in adults. Nevertheless, the paediatric dosage schedule for PCP was published in the label and leaflet insert at that time and continues so today.
As the first AIDS patients were adults, treated by doctors accustomed to treating adults and not children, many were prescribed TMP-SMZ, per leaflet insert, on the body weight basis, without recognition of a fixed adult dose [34]. Thus, the average 70 kg (154 lb ) man would receive a dose of 1400 mg TMP- 7000 mg SMZ per day, some 4.4 times the usual adult dose. To what extent these high doses given daily for two to three weeks have on adverse effects is not known. However, causes other than high dose were also at play. An increase in adverse reactions to pentamidine also occurred in AIDS. It is conceivable, but never proven, that one factor in the high success rate of TMP-SMZ for the prevention and treatment of PCP is the unusually high doses used.
3. Aerosolized Pentamidine
Unrelated to studies at St. Jude, an ingenious approach to the use of pentamidine devised in the late 1980s will be mentioned due to its importance. Because Pneumocystis organisms are located exclusively in the alveoli of the lung, with rare exception, Bernard, et al [35] Debs, et al [36], and Girard, et al [37] conceived the delivery of pentamidine by aerosol route into the lungs to avoid toxicity from systemic administration and they demonstrated feasibility in the rat model. Soon thereafter, clinical trials by Conte, et al [38] and Montgomery, et al [39] applied the method to patients with mild to moderate PCP and found favourable results. Later, Conte et al [40] and Soo Hoo, et al [41] found failure rates of 45 percent or greater. While this method is not recommended for treatment of PCP it has been shown to be effective and safe for prophylaxis in AIDS patients [42,43] and aerosolized pentamidine (NebuPent™) was approved by the FDA in 1989 for prophylaxis of PCP. In the multicentre controlled study of Schneider, et al 215 HIV-infected patients at high risk (CD4 cell count < 200 per cubic ml) for PCP were randomized to receive aerosolized pentamidine once a month or TMP-SMZ once a day [44]. Of the 71 patients receiving aerosol pentamidine, 6 (11%) had PCP, whereas none of the 142 patients on TMP-SMZ had PCP (P = 0.002). However, adverse events occurred more frequently in the TMP-SMZ than the pentamidine group.
4. Diaminodiphenylsulfone (Dapsone)
In 1981 St. Jude Children’s Research Hospital was far from the AIDS epidemic; located, not on east or west coasts, but in AIDS-free and PCP-free mid-America and served a clientele of infants and children, not adults. However, because of the more extensive use of TMP-SMZ possibly leading to drug resistance, and because some AIDS patients were showing intolerance to the drug, the infectious diseases investigators at St. Jude set out to find alternative drugs for PCP.
The cortisone/ prednisone-tetracycline-treated rat model was used again to screen drugs. Primaquine, chloroquine, suramin, difluoromethylornithine, allopurinol, ketoconazole, diloxanide, nifurtimox, melarsoprol, gentian violet and others were ineffective; whereas, diaminodiphenylsulfone (Dapsone) was highly effective in dose-dependent studies in rats (Table 7) [45].
Table 7 Comparison of three dose levels of dapsone with TMP-SMZ administered prophylactically in cortisone-treated rats [45].
|
Drug |
Dose, mg/kg/day |
No. (%) Rats with PCP |
|
None (control) |
0 |
9/9 (100%) |
|
Dapsone |
5 |
6/10 (60%) |
|
Dapsone |
25 |
0/10 (0%) |
|
Dapsone |
125 |
0/10 (0%) |
|
TMP-SMZ |
50-250 |
0/10 (0%) |
Further experiments showed that a combination of trimethoprim and dapsone was more effective than either of the drugs alone. Thus, trimethoprim is synergistic with dapsone, as it is with sulfamethoxazole.
Dapsone, a sulfone, was selected for testing because of its similar chemical structure to sulphonamides, its already established use in humans, low cost, reasonable safety and long plasma half-life (14 to 53 hours). Lacking local cases of PCP for study the St. Jude group was now limited in capability for clinical trials. Fortunately, colleagues at San Francisco General Hospital undertook the first clinical trials [46,47,48]. In a double-blind trial 60 patients with AIDS and PCP were randomized to receive TMP-SMZ or TMP-dapsone for treatment. The treatment was successful in 27 (90 %) of the 30 patients receiving TMP-SMZ and in 28 (93 %) of the 30 patients given TMP-dapsone. Furthermore, there were fewer serious adverse reactions with TMP-dapsone than with TMP-SMZ. Subsequent studies showed dapsone was less effective than TMP-dapsone [46], as predicted by the rat model studies [45]. Most of the AIDS patients intolerant to TMP-SMZ were able to take TMP-dapsone.
The long plasma half-life (up to 53 hours), relative safety, and low cost suggested that dapsone given at the infrequent dose of once weekly might be effective in PCP prophylaxis and would be especially applicable in underdeveloped countries and in circumstances where supervised administration was practiced. Several studies showed this to be the case [49,50,51]. Dapsone and trimethoprim-dapsone were included in the CDC, NIH, and Infectious Diseases Society of America Guidelines for the treatment and prevention of PCP in patients with HIV/AIDS and continue today as an alternative to TMP-SMZ. Dapsone and pyrimethamine plus leucovorin is recommended for patients seropositive for Toxoplasma gondii who cannot tolerate TMP-SMZ [15].
5. Atovaquone (Mepron™ = trans-2-[4-(4-chlorophenyl) cyclohexyl]-3-hydroxy-1, 4-naphthalenedione)
The St. Jude investigators were especially anxious to find a drug that had its target other than the folate pathway (trimethoprim inhibits dihydrofolate reductase and sulfamethoxazole inhibits dihydropteroate synthase by competing with para-aminobenzoic acid) [52]. By the late 1980s, TMP-SMZ had been given to several million people worldwide for long courses of time and it was feared that drug-resistant strains of Pneumocystis species might arise or that drug-resistant bacteria from the microbial flora might become troublesome. Also, a non-antifol might be useful for patients having adverse effects from TMP-SMZ, dapsone and pentamidine. Serendipitously, a fortuitous set of events led to the discovery and development of such a drug.
Totally unrelated to Pneumocystis and HIV/AIDS, scientists at the Wellcome Research Laboratories in Beckenham, England were beginning to investigate a new antimalarial compound, 2-[trans-4-(4-chlorophenyl)cyclohexyl]-3-hydroxy-1,4-naphthoquinone, designated as 566C80. In search of other activities that might expand the use of the new drug, Dr. Win Gutteridge contacted the St. Jude group as to our interest in testing 566C80 for anti-PCP efficacy in the rat model. Learning the hydroxynaphthoquinone, a structural analogue of ubiquinone, probably blocks the electron transport chain, indirectly inhibits the activity of dihydroorotate dehydrogenase, and stops the de novo synthesis of pyrimidines in Plasmodium falciparum, we undertook screening in the cortisone-treated rat model for anti-PCP activity. The 566C80 compound was found to be highly effective (100 %) in doses of >100 mg/kg/day (Table 8), with no signs of adverse effects in animals [10,53]. Later, Gutteridge and his group repeated the experiments in England and found the same results [10].
Table 8 Rat experiments: Extent of PCP after prophylaxis with 566C80 (atovaquone) at various dose levels [10].
|
Drug |
Dose (mg/kg/day) |
No. Rats |
% with PCP |
|
Control (no drug) |
none |
10 |
100% |
|
TMP-SMZ |
50/250 r |
10 |
0% |
|
566C80 (atovaquone) |
200 r |
10 |
0% |
|
566C80 |
100 r |
10 |
0% |
|
566C80 |
100 g |
10 |
10% |
|
566C80 |
50 g |
10 |
30% |
|
566C80 |
25 g |
10 |
90% |
|
566C80 |
10 g |
10 |
90% |
r = drug in rations (“with food”), g = administered by gavage in fasting state (“without food”).
To evaluate 566C80 further, Wellcome Research Laboratories in UK and its sister company Burroughs Wellcome Co. at Research Triangle Park in U. S. collaborated with St. Jude Children’s Research Hospital for a Phase 1 study in HIV-infected men (>18 yrs.) [11]. Twenty-five subjects in cohorts of five men received total daily doses of 100, 250, 750, 1500, and 3000 mg orally in tablet form for 12 days. The only drug-related adverse event was a maculopapular rash in one patient that resolved while the drug was continued. With the highest dose of 3000 mg, pharmacokinetic values showed the maximum plasma concentration = 39 µg/ml; time to maximum plasma concentration = 8 h; area under plasma concentration-time curve at steady state = 1088 h·µg/ml; plasma half-life = 51 h and total plasma clearance = 4.09 l/h. Since the absorption of 566C80 was increased in the rat studies when given with food, attention was given to optimize food administration in the human study. It is important to note that two- to threefold higher plasma concentrations are achieved when the drug is given with food than when fasting, a point important to clinical practice.
A multicentre Phase I - Phase II open-label, dose-escalation study of the treatment of mild-to-moderately severe PCP in 34 adults with AIDS was undertaken by Falloon, et al in 1991 [12]. All of the 34 patients survived the PCP, with 27 (79%) receiving 566C80 alone. The drug was stopped in 4 (12%) patients because of possible adverse effects (fever in 2, rash in 2).
The pivotal study to gain FDA approval required a comparison of 566C80 and TMP-SMZ in the treatment of PCP. Despite the rampant worldwide spread of AIDS, the number of cases of PCP was limited at any single medical centre or city because of widespread PCP prophylaxis and more effective antiretroviral therapy. The phase III study was designed as a randomized, double-blind, multicentre trial with enough subjects with PCP and AIDS to establish that 566C80 (now given the generic name atovaquone) was no less successful therapeutically than the currently recommended TMP-SMZ [13]. Because of the high efficacy rate of TMP-SMZ it was deemed unachievable to power the study with enough patients to determine if 566C80 (atovaquone) might be more effective than TMP-SMZ. Thirty-seven medical centres in the United States, Canada, Belgium, France, Germany, England and The Netherlands participated in the trial; and were aided by the National Institutes of Health, AIDS Clinical Trials Groups.
Of 322 AIDS patients with histologically confirmed PCP, 160 were randomized to receive atovaquone (750 mg) and 162 to receive TMP (320 mg)-SMZ (1600 mg) three times daily for 21 days. The results are summarized in Table 9. Therapy involving only the initial drug was successful and free of adverse effects in 62 percent of those receiving atovaquone and 64 percent of those given TMP-SMZ. Treatment-limiting adverse effects required a change of drugs in 7 percent of the atovaquone group and in 20 percent of the TMP-SMZ group (Table 10). Evaluation for therapeutic efficacy showed success in 80 percent of those on atovaquone and 93 percent of those on TMP-SMZ. In sum, there was a trade-off with more efficacy from TMP-SMZ for more safety with atovaquone [13,54]. The safety factor of atovaquone makes it a favourable alternative for patients having adverse reactions to TMP-SMZ.
Table 9 Comparison of atovaquone (566C80) with trimethoprim-sulfamethoxazole (TMP-SMZ) to treat Pneumocystis carinii pneumonitis in patients with AIDS [13].
|
Drug |
Number |
Efficacy |
Treatment Limiting |
Therapy |
|
Patients |
Success* |
Adverse Events** |
Success* |
|
|
Atovaquone tablets |
160 |
80% |
7% |
62% |
|
TMP-SMZ |
162 |
93% |
20% |
64% |
*P = 0.002, ** P = <0.001
Table 10 Treatment-limiting adverse effects among all patients enrolled in the comparison study [13].
|
Adverse Effect |
Frequency |
Exact P Value |
|
|
No. (%) of Patients |
|||
|
Atovaquone (n= 203) |
TMP-SMZ (n= 204) |
||
|
Rash |
8 (4) |
16 (8) |
0.14 |
|
Liver dysfunction |
5 (3) |
15 (7) |
0.04 |
|
Vomiting |
2 (1) |
14 (7) |
<0.01 |
|
Fever |
1 (1) |
13 (6) |
<0.01 |
|
Nausea |
1 (1) |
11 (5) |
<0.01 |
|
Neutropenia |
0 (0) |
7 (3) |
0.02 |
|
Pruritis |
1 (1) |
3 (2) |
0.63 |
|
Chills |
0 (0) |
3 (2) |
0.25 |
|
Headache |
0 (0) |
3 (2) |
0.25 |
|
Renal impairment |
0 (0) |
3 (2) |
0.25 |
|
Thrombocytopenia |
0 (0) |
3 (2) |
0.25 |
|
All |
19 (9) |
50 (24) |
<0.01 |
Mean steady-state plasma concentrations were determined for atovaquone and TMP-SMZ during the course of treatment for PCP. Logistic-regression analyses showed the probability of a therapeutic response was strongly associated with the plasma concentrations of atovaquone (Figure 2). The outcome was successful for 96.7 % of patients with steady-state concentrations of 15µg/ml or greater of atovaquone. There was a direct relation between therapeutic success and the plasma concentration of the drug, a point of importance suggesting that if higher concentrations can be achieved, a more effective therapeutic outcome might be expected without increasing adverse effects. It is noteworthy that the original tablet formulation was used in this study and later a liquid formulation was developed that provided better absorption and higher plasma concentrations. With TMP-SMZ the therapeutic success and incidence of treatment-limiting events were not related to plasma concentrations of either of the drug combination, possibly because concentrations were well above what was needed.
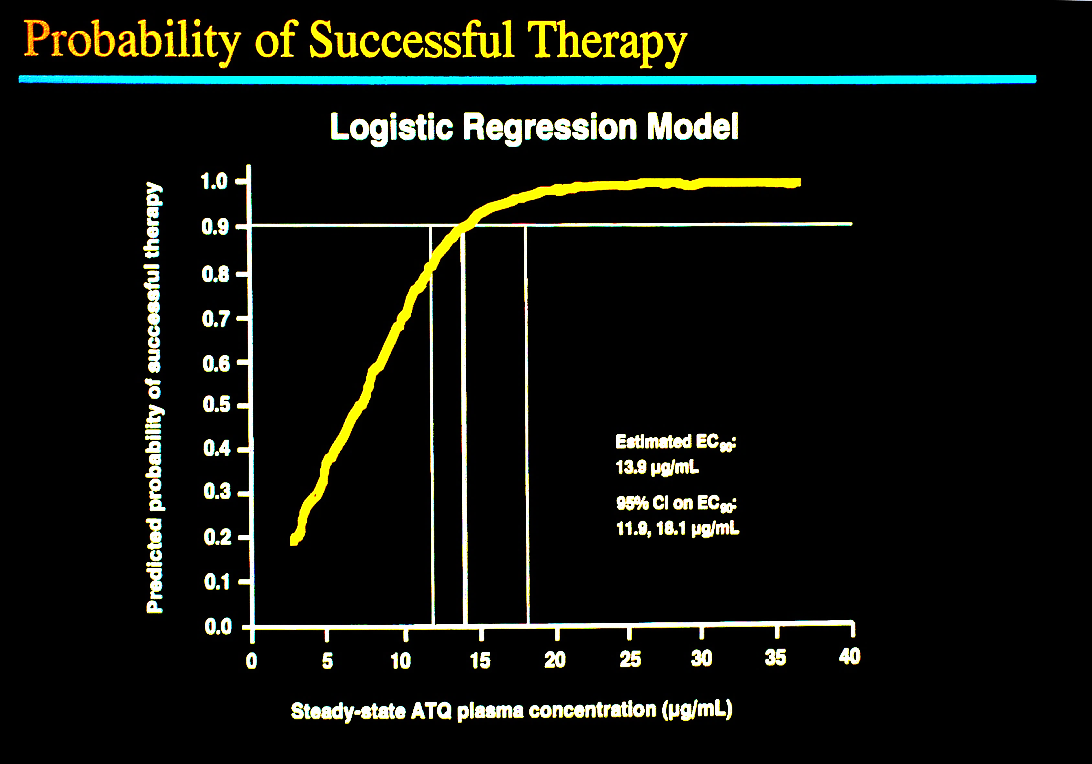
Figure 2 Relationship of steady-state concentration of atovaquone and therapeutic success for PCP in patients with AIDS [13,53]. The graph shows a direct relationship of the plasma concentration and successful treatment of PCP.
The FDA approved the tablet formulation of 566C80 (atovaquone) in 1993 when it was given the brand name Mepron™. The pharmacologists at Burroughs-Wellcome/ Glaxo soon devised a micronized liquid formulation (m-atovaquone) that provided better absorption and higher plasma concentrations of atovaquone. The FDA approved the suspension formulation in January 1999 and the tablet formulation was discontinued. Note that all the clinical trials described above were done with the tablet form of atovaquone [11,12,13].
A phase 1 study in children showed doses of 30 to 45 mg/kg/day of the new suspension of m-atovaquone provided average steady-state concentrations well above the 15 µg/ml level shown in earlier studies for successful outcome in treatment 96.7 % of PCP cases [55].
Eighty-six children with leukaemia and intolerant to TMP-SMZ at St. Jude Children’s Research Hospital were given daily atovaquone suspension for a total of 172.1-person years and no cases of PCP occurred [56].
El-Sadr, et al [57] conducted a multicentre, open-label, randomized trial comparing atovaquone suspension 1500 mg per day with dapsone, 100 mg per day for the prevention of PCP among 1057 patients with HIV infection who were intolerant to TMP-SMZ. After an average observation of 27 months results showed atovaquone and dapsone were similarly effective in the prevention of PCP, but atovaquone was better tolerated and was the preferred choice for PCP prophylaxis.
Unlike TMP-SMZ, atovaquone has no antibacterial effects, although it is effective in the treatment of malaria, toxoplasmosis and babesiosis [58]. Because most of the immunocompromised hosts at risk for PCP are also at risk for bacterial infections and benefit from broad-spectrum antimicrobial prophylaxis of TMP-SMZ, the antibiotic azithromycin was combined with atovaquone and compared to TMP-SMZ in 366 children with HIV who qualified for PCP prophylaxis [59]. The randomized, double-blind, placebo-controlled trial showed that the rates of serious bacterial infections were lower among the azithromycin-atovaquone recipients than TMP-SMZ, and the groups were similar as to PCP breakthrough rates, non-bacterial infections and adverse event profiles. Statistically, the results marginally favoured azithromycin-atovaquone prophylaxis.
The cost of atovaquone suspension (Mepron™) has been a significant factor in its usage. It is more expensive than any of the other regimens for PCP prophylaxis.
6. Aerosolized Atovaquone and Surfactant
PCP is unique among infections of the immunosuppressed host in that the organism resides only in the lower airways of the lung and its presence in the alveolar spaces allows it to be continually bathed directly in inhaled air. Also, the disease it causes is limited to this organ, even in fatal cases. The entity is ideal for delivery of therapeutic agents via the inhalation route. This route has been used extensively in the treatment of asthma and other chronic lung diseases, as well as aerosolized pentamidine for the prevention of PCP. Separately, for several decades Paediatricians have administered surfactant down the airways of new-born infants with acute respiratory distress syndrome (hyaline membrane disease) with remarkable life-saving success. These factors led the investigators at St. Jude to undertake a series of experiments with the cortisone-treated rat model to evaluate the use of aerosolized atovaquone and synthetic surfactant in combating PCP [60]. Earlier studies had shown diminished phospholipids of surfactant in rats [61,62,63,64,65] and humans [66,67] with PCP. Normally, the type II alveolar lining cells secrete the phospholipid-protein complex surfactant onto the surfaces of the alveolar wall. This stabilizes the alveoli at the end of respiration, reduces the surface tension at the air-liquid interface, and reduces the energy required for breathing. In the experiments to follow it was hypothesized that the administration of synthetic surfactant and atovaquone by aerosol, either separately or in combination would achieve an improved antimicrobial effect as well as an improvement of oxygenation and pulmonary function.
Aerosol delivery devices shown in Figure 3 provided accurate delivery of nebulized atovaquone (10.12 mg/ml) and synthetic surfactant (colfosceril palmitate 81.0 mg/ml) to the lungs of experimental rats under a variety of conditions to determine the pharmacokinetics and efficacy in the prevention and treatment of PCP [60].
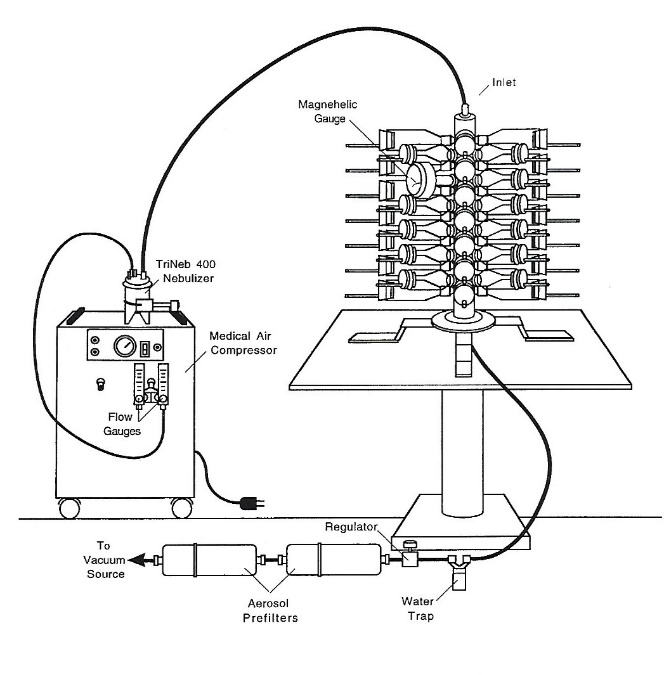
Figure 3 Cannon Flow-Past Nose-Only Exposure System (Lab Products. Maywood, NJ) used to deliver aerosols of atovaquone and surfactant to rats with PCP [60].
After a single dose of aerosolized atovaquone the mean peak concentrations of atovaquone averaged 52µg/mL in plasma and 31µg/g in lungs of rats infected with P. carinii (Figure 4). The EC100 concentration that prevented PCP in 100 percent of rats was > 1.28; < 1.78 mcg/g of lung (Figure 5). When atovaquone was combined with surfactant mean peak concentrations of 94µg/ml in plasma and 51µg/g in lung were obtained.
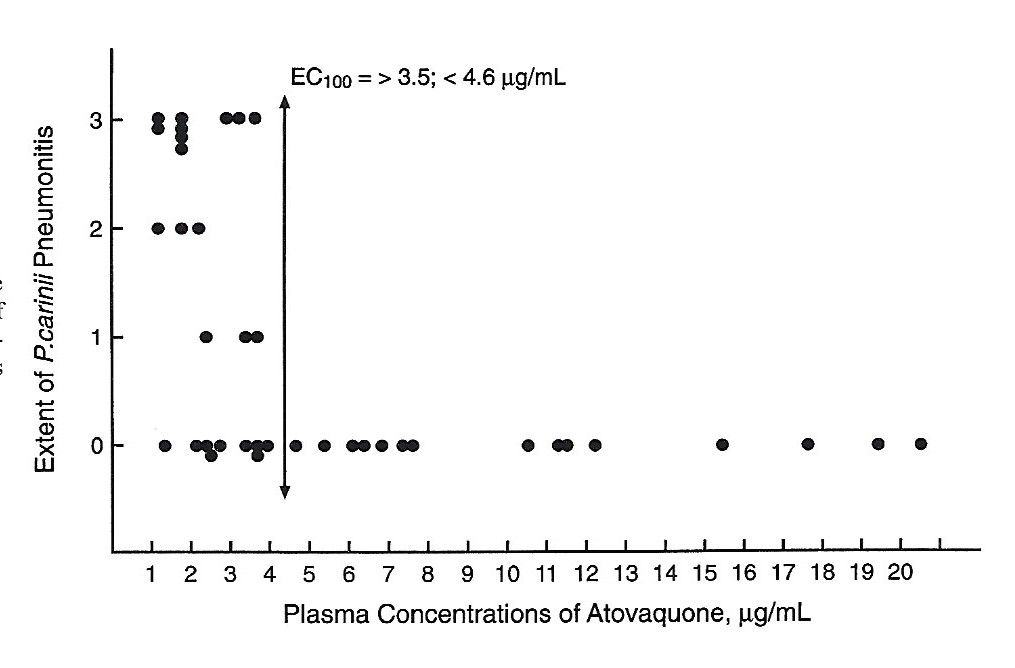
Figure 4 Comparison of atovaquone concentrations in plasma and occurrence of PCP. EC100 = the concentration that prevented PCP in 100 % of rat.
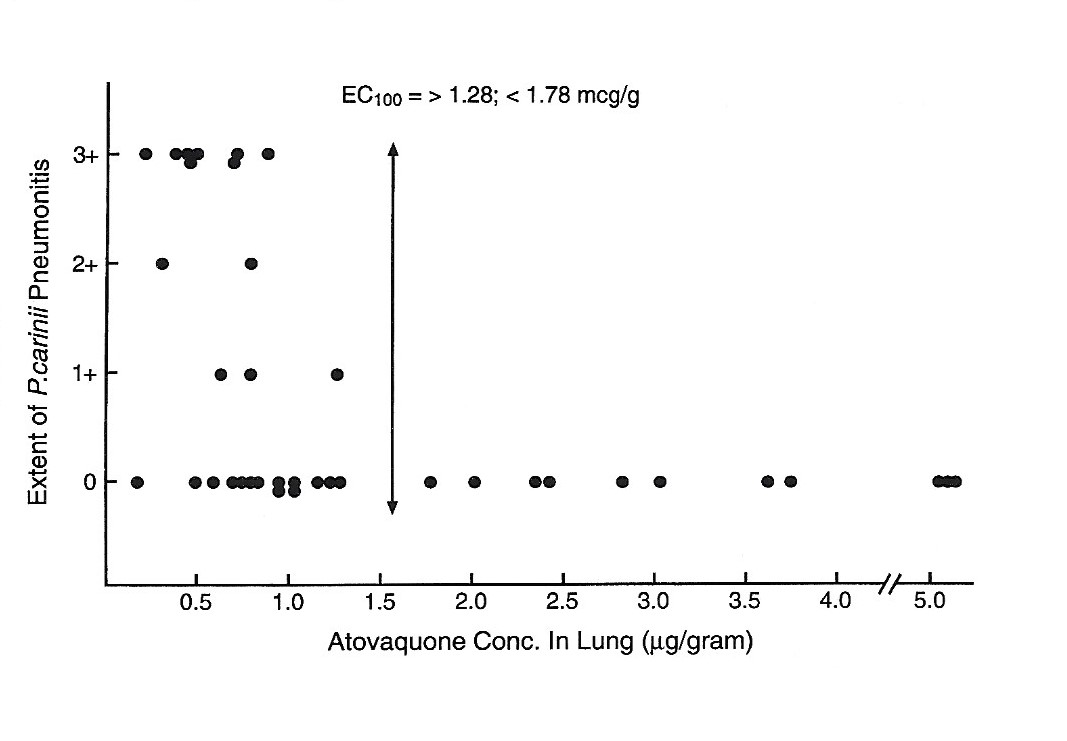
Figure 5 Comparison of atovaquone concentrations in lungs and the occurrence of PCP.
An impressive finding was observed when aerosolized surfactant was administered alone, without antimicrobial drugs. As shown in Figure 6 the survival of rats with PCP was greatly increased. When atovaquone was combined with surfactant the concentrations of atovaquone were increased beyond those with atovaquone alone. By day 70 of cortisone immunosuppression 14 (61 percent) of the 23 rats receiving surfactant were surviving compared to 1 (5 percent) of the 21 control animals.
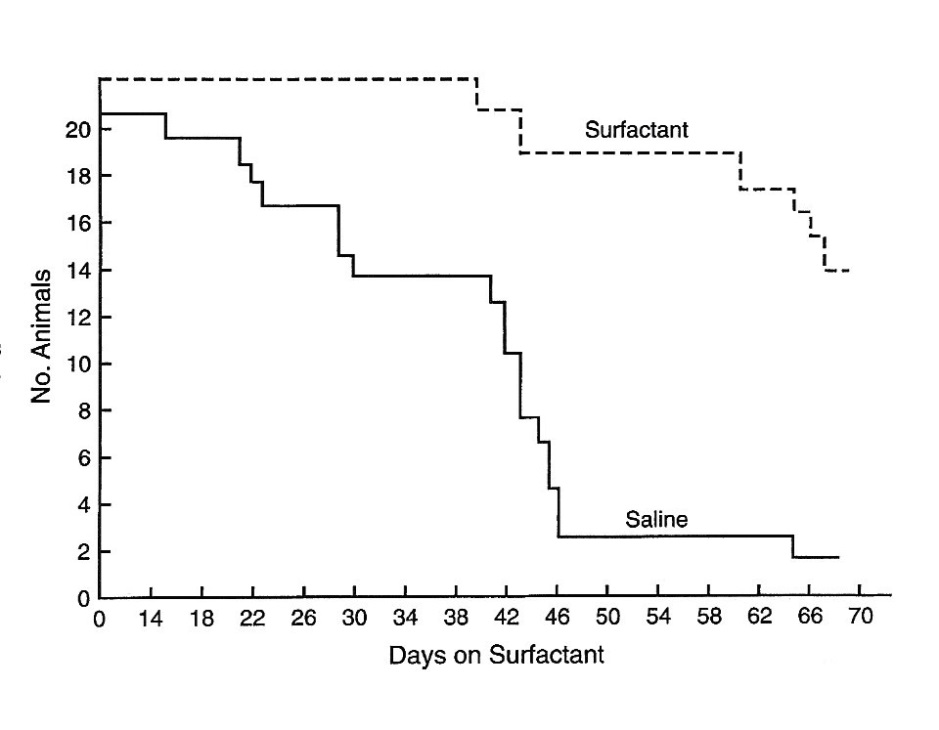
Figure 6 Kaplan-Meir survival plot of cortisone-treated rats with PCP maintained prophylactically on surfactant or saline aerosols [60].
Aerosolized atovaquone administered once weekly with or without surfactant prevented PCP, but oral administration once a week was inadequate to prevent PCP. Histopathological examination of the lungs revealed no apparent evidence of damage due to the drugs.
The next step intended for this study was the development of a hand-held, self-delivery nebulizing device for the administration of an adequate dose of atovaquone suspension for adults, followed by a phase I study for safety and pharmacokinetics of aerosol atovaquone with or without surfactant.
7. Opportunities for Future Clinical Trials to Advance PCP Therapeutics
If a plea is to come from these pages, it will be for more vigorous and productive translational research on the prevention and treatment of infection from Pneumocystis jirovecii in humans. Despite an abundance of basic research on Pneumocystis sp. during the past several decades, little has been achieved for new major advancements in the clinical management of patients with or at risk for PCP. In fact, the standard methods for diagnosis, prevention and treatment for the practicing physician are essentially those in use a quarter century ago. The main problem is not so much a need for more intensive research in the laboratory for new drugs, although that is always welcome; the barrier is a dearth of quality translational trials to advance drugs from the laboratory into clinical practice. Unless a new compound is taken from the laboratory to the clinic and qualified for use in humans, the labours of the scientists in drug discovery have been essentially for naught. In addition to the dozens of interesting and promising compounds that have been discovered in recent years, the availability of the P. jirovecii genome sequences provides a grand opportunity for continued drug discovery of enormous dimensions. Nevertheless, the existing arsenal of drugs already in the lab offers a useful resource for the clinical investigator today.
7.1. New Anti-Pneumocystis jirovecii Drugs
Cushion and Walzer provide a comprehensive review of candidate anti-P. jirovecii drugs identified and developed by contemporary investigators [68]. References are provided in their article for antifolate compounds, including dihydropteroate synthase inhibitors (sulphonamides, sulfones and sulfonylureas) alone or in combination with inhibitors of dihydrofolate reductase; macrolides combined with sulphonamides; diamidines and related cationic compounds; 8-aminoquinoliones with and without clindamycin; purine nucleoside analogues; polyamine inhibitors; nitrofurans; papulocandins; echinocandins; sodarins; allylamines; streptogrammins; fluoroquinolones; hydroxynaphthoquinolones with or without dihydrofolate reductase inhibitors, macrolides, or rifamycins; ionophores; benzonaphthacenes; iron chelators; immunological agents; and others with novel mechanisms of action. Since their review even more new drugs have been reported, not covered in my commentary which is limited to studies from our centre.
Mention can be made of the echinocandins as an example of the premise of my commentary, that is, the need for proper clinical trials of promising compounds found to be effective against Pneumocystis sp. in the laboratory. Since the original and convincing report of Schmatz, et al [69] in 1990 of the efficacy of an echinocandin and a papulocandin against PCP in the rat model numerous reports of laboratory studies, e.g. Cushion, et al [70], Lobo, et al [71], Aliouat, et al [72], Gomes, et al [73] and others, have generally supported the effectiveness of these 1, 3-β-glucan synthesis inhibitors. However, during this period of more than 25 years no clinical trial has been undertaken to prove or disprove efficacy in the treatment or prevention of PCP in humans. Meanwhile the echinocandin Caspofungin was evaluated in clinical trials for the treatment of candidiasis and aspergillosis, became approved by the FDA for this purpose (not for PCP) and has been used extensively in medical practice. Intravenous infusion is required and adverse reactions are common. Case reports from “off label” use of the echinocandin for PCP have been reviewed recently (2018) by Huang, et al [74] and earlier by Armstrong, et al [75] with the conclusions that the drug may have efficacy, especially if combined with trimethoprim-sulfamethoxazole, and that a proper controlled clinical trial is needed for definitive results.
After the onset of the AIDS epidemic the St. Jude investigators spent more than a decade in the laboratory searching for new drugs for use in the prevention and treatment of PCP. Their studies identified several highly effective drugs in the animal model, including sulfonylurea compounds [76]; 4, 4’ sulfonylbisformaldehyde [77]; synergy of macrolide + sulfonamides [78,79] ; PS-15 (new biguanide) [80]; mono-sulfonamides [81]; mycophenolate mofetil [82]; lasalocid [83]; and aerosol atovaquone [60]. None of these drugs, nor the promising concept of aerosolized atovaquone have been brought to clinical trials; so, their efforts, plus many years of N. I. H. treasure, have been a total waste. One must now question the wisdom of accumulating huge amounts of anti-PCP drugs that will never be applied to humans. This tongue-in-cheek statement is not intended to thwart future brilliant scientific research in the laboratory but rather to foster accelerated translational studies in the clinic.
Several difficult problems face the clinical investigator today seeking to contribute new modalities to the prevention and treatment of PCP. While the pneumonitis is increasing in prevalence worldwide, it is sparsely distributed and in patients with another disease and the underlying disease may be designated for anti-PCP prophylaxis as a standard practice, precluding enrolment in some PCP protocols. Therefore, few medical centres can accrue enough patients within a reasonable time to undertake a clinical trial to determine endpoints with sound statistical analysis. Furthermore, statisticians are now pushing to change their magical P value from 0.05 to 0.005 to claim statistical significance, possibly requiring an increase in bulk to population samples [84]. In most instances clinical trials beyond Phase I studies will require multicentre collaboration. On the positive side recent changes in Federal regulations have allowed more rapid processing of applications by the FDA and the establishment of “Right to try” opportunities to enhance screening of some new drugs. Innovative clinical investigators should take advantage of these opportunities.
7.2. Adverse Reactions to Drugs
There is no greater morass in clinical research than the determination of adverse events due to therapeutic drugs. PCP is a prime example. A common call to justify new drugs for PCP is “because of the high rates of adverse events caused by drugs in use.” Herein lies the rub. Investigators in clinical trials are required to identify and report adverse events with FDA submissions from phase 1 through subsequent studies. After FDA approval, post marketing surveillance continues through the FDA Adverse Event Reporting System (FAERS). Any abnormal event encountered during the clinical trial that cannot be otherwise explained is considered an “adverse event” to the experimental drug. Although the system is reasonably effective in identifying all the possible adverse effects of study drugs, it does little to prove that the presumed adverse event is caused by the drug under study. At the time of licensure, the FDA prepares an official label and package insert listing all adverse events reported from clinical trials. This information is the resource for practitioners and patients in considering adverse events to specific drugs. Post-marketing reports from patients and physicians of adverse events continues to accrue more and more entities through FAERS. In practice investigators seldom attempt or, if so, succeed in validating that the adverse event is caused by the drug and not another cause.
As an example of the dilemma is the entry for TMP-SMZ, considered a relatively safe antibiotic, in the current FDA package insert [85]:
“ADVERSE REACTIONS: nausea, vomiting, anorexia, rash, Steven-Johnson syndrome, toxic epidermal necrolysis, fulminant hepatic necrosis, agranulocytosis, aplastic anaemia, thrombocytopenic, leukopenia, neutropenia, haemolytic anaemia, megaloblastic anaemia, hypothrombinaemia, methemoglobinemia, eosinophilia, anaphylaxis, allergic myocarditis, erythema multiforme, exfoliative dermatitis, angioedema, drug fever, chills, Henoch-Schoenlein purpura, serum sickness-like syndrome, generalized allergic reaction, generalized skin eruptions, photosensitivity, conjunctival and scleral injection, pruritis, urticaria, periarteritis nodosa, systemic lupus erythematosus, hepatitis, elevation of serum transaminase and bilirubin, pseudomembranous enterocolitis, pancreatitis, stomatitis, glossitis, emesis, abdominal pain, diarrhoea, renal failure, interstitial nephritis, BUN and creatinine elevation, toxic nephrosis with oliguria and anuria, crystalluria and nephrotoxicity in association with cyclosporine, hyperkalaemia, aseptic meningitis, convulsions, peripheral neuritis, ataxia, vertigo, tinnitus, headache, hallucinations, depression, apathy, nervousness, diuresis, hypoglycaemia, arthralgia, myalgia, rhabdomyolysis, weakness, fatigue, insomnia, fever, cough, shortness of breath, pulmonary infiltrates.”
How ridiculous! If the list is followed, one would find that 100 percent of patients with PCP would have adverse events related to TMP-SMZ because they have fever, cough, shortness of breath, and pulmonary infiltrates listed above [85].
There is an analogy from the 19th century to the current adverse event quagmire. At the time when attempts were being made to establish the etiology of microbial diseases, any bacterium isolated from any ill patient was considered the cause of the disease. It wasn’t until Robert Koch proposed a set of strict “postulates” to better define the relationship that the true etiology of a disease was delineated. Although the famous Koch’s Postulates are no longer touted, a similar approach might help deal with the morass of adverse effects from pharmaceuticals if investigators applied them in the design of clinical trials. Some examples are suggested [86].
Postulate 1: The adverse event is temporally related to administration of the drug.
Postulate 2: The same adverse event recurs when the patient is re-challenged with the drug.
Postulate 3: The adverse event occurs in the absence of other drugs and underlying diseases, known to have similar clinical or laboratory- defined reactions.
Postulate 4: The adverse event, if at a reversible stage, resolves when the drug is discontinued.
Postulate 5: The vehicle in which the drug is prepared does not cause the adverse event.
Postulate 6: Toxicity is dose dependent.
Postulate 7: Adverse events attributed to drug allergy or hypersensitivity should fulfil the patterns of types I, II, III or IV allergic reactions [86].
A point to be made here is that the experimental design of clinical trials for new drugs in the treatment and prevention of PCP should include stringent assessments to accurately identify all adverse reactions from the drug under consideration, and to also assure that the drug is not accused of reactions due to other causes.
Success in many areas of medicine now allows patients to survive longer and in better states of health but at the expense of drug usage that impairs immunity. Such is the case with cancer, organ transplantation, AIDS/HIV infection, congenital and acquired immunodeficiency disorders, and some autoimmune disorders. Recently, PCP prophylaxis has been recommended for certain patients with rheumatoid arthritis (RA), a chronic disease that affects between one and two million patients in the United States alone [87]. Thus, the problems of Pneumocystis jirovecii will persist. The future with scholarly studies of Pneumocystis sp. in the laboratory and the clinic offers great promise and excitement.
Author Contributions
The author made all contributions to this work.
Competing Interests
The authors have declared that no competing interests exist.
References
- Hughes WT, Kuhn S, Chaudhary S, Feldman S, Verzosa M, Aur RJA, et al. Successful chemoprophylaxis for Pneumocystis carinii pneumonitis. N Engl J Med. 1977; 297: 1419-1426. [CrossRef]
- Hughes WT. Pneumocystis carinii pneumonia. N Engl J Med. 1977; 297: 1381-1383. [CrossRef]
- Hughes WT, McNabb PC, Makres TD, Feldman S. Efficacy of trimethoprim and sulfamethoxazole in the prevention and treatment of Pneumocystis carinii pneumonitis. Antimicrob Agents Chemother. 1974; 5:289-293. [CrossRef]
- Hughes WT, Feldman S, Sanyal SK. Treatment of Pneumocystis carinii pneumonitis with trimethoprim-sulfamethoxazole. Canad Med Assoc J. 1975; 112: (13 spec no.) 47s-50s.
- Hughes WT, Feldman S, Chaudhary SC, Ossi MJ, Cox F, Sanyal SK. Comparison of pentamidine isethionate and trimethoprim- sulfamethoxazole in the treatment of Pneumocystis carinii pneumonia. J Pediatr. 1978; 92: 285-291. [CrossRef]
- Hughes WT. Treatment of Pneumocystis carinii pneumonitis. N Engl J Med. 1976; 295: 726-727. [CrossRef]
- Hughes WT, Rivera GK, Schell MJ, Thornton D, Lott L. Successful intermittent chemoprophylaxis for Pneumocystis carinii pneumonia. N Engl J Med 1987; 316: 1627-1632. [CrossRef]
- Hughes WT, Smith BL. Efficacy of diaminodiphenylsulfone and other drugs in murine Pneumocystis carinii pneumonitis. Antimicrob Agents Chemother. 1984; 26: 436-440. [CrossRef]
- Mills J, Leoung G, Medina I, Hopewell PC, Hughes WT, Wofsy C. Dapsone treatment of Pneumocystis carinii pneumonia in the acquired immunodeficiency syndrome, Antimicrob Agents Chemother. 1988; 32: 1057-1060. [CrossRef]
- Hughes WT, Gray V, Gutteridge WE, Latter VS, Pudney M. Efficacy of a hydroxynaphthoquinone (566C80) in experimental Pneumocystis carinii pneumonia. Antimicrob Agents Chemother. 1990; 34: 225-228. [CrossRef]
- Hughes WT, Kennedy W, Shenep JL, Flynn PM, Hetherington SV, Fullen G, et al. Safety and pharmacokinetics of 566C80, a hydroxynaphthoquinone with anti-Pneumocystis carinii activity: a phase 1 study in human immunodeficiency virus (HIV)-infected men. J Infect Dis. 1991; 163: 843-848. [CrossRef]
- Falloon J, Kovacs J, Hughes WT, O’Neill D, Polis M, Lancaster D, et al. A preliminary evaluation of 566C80 for the treatment of Pneumocystis carinii pneumonia in patients with acquired immunodeficiency syndrome. N Engl J Med. 1991; 325: 1534-1538. [CrossRef]
- Hughes WT, Leoung G, Kramer F, Bozzette S, Safrin S, Frame P, et al. Comparison of atovaquone (566C80) with trimethoprim-sulfamethoxazole for the treatment of Pneumocystis carinii pneumonia in patients with AIDS. N Engl J Med. 1993; 328: 1521-1527. [CrossRef]
- Ivady G, Paldy L. Ein neues Behandlungsverfahren der interstitiellen plasmazelligen Pneumonie Fruhgeborener mit funfwertigem Stibium und aromatischen Diamidinen. Monatsschr Kinderheildk. 1958; 106: 10-16.
- Panel on Opportunistic Infections in HIV-infected Adults and Adolescents. Guidelines for the prevention and treatment of opportunistic infections in HIV-infected adults and adolescents: recommendations from the Centres for Disease Control and Prevention, the National Institutes of Health, and the HIV Medicine Association of the Infectious Diseases Society of America. Available at http://aidsinfo.nih.gov/contentfiles/adult_oi.pdf. Accessed 7/23/2018.
- Perera DR, Western K, Johnson HD, Johnson WW, Schultz MG, Agers PV. Pneumocystis carinii pneumonia in a hospital for children. Epidemiological aspects. JAMA 1970; 214: 1074-1078. [CrossRef]
- Hughes WT, Price RA, Kim HK, Coburn TP, Grigsby D, Feldman S. Pneumocystis carinii pneumonia in children with malignancies. J Pediatr 1973; 82: 404-415. [CrossRef]
- Weller R. Production of pneumocystosis in animals. Z Kinderchir. 1955; 76: 366-378.
- Weller R. Further studies of experimental pneumocystosis of rats with regard to interstitial pneumonia of premature infants. Z Kinderchir. 1956; 78: 166-176.
- Frenkel JK, Good JT, Shultz JA. Latent Pneumocystis carinii infection of rats, relapse and chemotherapy. Lab Invest. 1966; 15: 1559-1577.
- Hughes WT, Kim HK, Price RA. Attempts at prophylaxis for murine Pneumocystis carinii pneumonitis. Curr Therap Res. 1973; 15 581-587.
- Kim HK, Hughes WT, Feldman S. Studies of morphology and immunofluorescence of Pneumocystis carinii. Proc Soc Exp Biol Med. 1972; 141: 304-309. [CrossRef]
- Springer JR, Beard CB, Miller RF, Wakefield AE. A new name for Pneumocystis from humans and new perspectives on host-parasite relationship. Emerg Infect Dis J. 2002; 8: 891-896. [CrossRef]
- Wilber RB, Feldman S, Malone WJ, Ryan M, Aur RJ, Hughes WT. Chemoprophylaxis for Pneumocystis carinii pneumonitis: outcome of unstructured delivery. Amer J Dis Child. 1980; 134:643-648. [CrossRef]
- Hughes WT. Five-year absence of Pneumocystis carinii pneumonitis in a pediatric oncology center. J Infect Dis. 1984; 150: 305-306. [CrossRef]
- Harris RE, McCallister JA, Allen SA, Burton AS, Baehner RL. Prevention of Pneumocystis carinii pneumonia. Use of continuous sulfamethoxazole-trimethoprim therapy. Amer J Dis Child. 1980; 134: 35-38. [CrossRef]
- Wolff LJ, Baehner RL. Delayed development of Pneumocystis carinii pneumonia following administration of short-term high-dose trimethoprim-sulfamethoxazole. Amer J Dis Child. 1978; 132: 525.
- Chusid MJ, Heyrman KA. An outbreak of Pneumocystis carinii pneumonia in a pediatric hospital. Pediatrics 1978; 62: 1031.
- Hughes WT. Pneumocystis carinii Pneumonia. CRC Press, Boca Raton. 1987. p. 266.
- Hughes WT, Smith BL. Intermittent chemoprophylaxis for Pneumocystis carinii pneumonia. Antimicob Agents Chemother. 1983; 24:300-301. [CrossRef]
- Masur H, Michelis MA, Greene JB, Onarato I, Stouwe RAV, Cunningham-Rundles S, et al. An outbreak of community acquired Pneumocystis carinii pneumonia: initial manifestations of cellular immune dysfunction. N Engl J Med. 1981; 305: 1431-1438. [CrossRef]
- Gottlieb MS, Schroff R, Schanker HM, Weisman JD, Fan PT, Wolf RA, et al. Pneumocystis carinii pneumonitis and mucosal candidiasis in previously healthy homosexual men: evidence of a new acquired cellular immunodeficiency. N Engl J Med. 1981; 305: 1424-1431. [CrossRef]
- Jaffee H, Choi K, Thomas PA, Haverkos HW, Auerbach DM, Gunan ME, et al. National case control study of Kaposi’s sarcoma and Pneumocystis carinii pneumonia in homosexual men: Part 1. Epidemiological results. Ann Intern Med. 1983; 99: 145-151. [CrossRef]
- Jaffee HS, Abrams DL, Lewis BJ, Golden JA. Complications of cotrimoxazole in the treatment of AIDS-associated Pneumocystis carinii pneumonia in homosexual men. Lancet 1983; 2: 1109-1111. [CrossRef]
- Bernard EM, Donnell HS, Huang A, Tsany A, Armstrong D. Successful prevention and treatment of experimental Pneumocystis carinii pneumonia with aerosolized pentamidine. In Abstracts of the 2nd International Conference on AIDS. Abstract A300, Paris, June, 1986.
- Debs RJ, Blumenfeld W, Burnette EJ, Straubinger RM, Montgomery AB, Lui E, et al. Successful treatment with aerosolized pentamidine in Pneumocystis carinii pneumonia in rats. Antimicrob Agents Chemother. 1987; 31: 37-41. [CrossRef]
- Girard PM, Brun-Pascaud M, Farinotti R, Tamisier L, Kernbaum S. Pentamidine aerosol in prophylaxis in murine Pneumocystis carinii pneumonia. Antimicrob Agents Chemother. 1987; 31: 978-981. [CrossRef]
- Conte JE, Hollander H, Golden JA. Inhaled or reduced dose intravenous pentamidine for Pneumocystis carinii pneumonia. Ann Intern Med. 1987; 107: 495-498. [CrossRef]
- Montgomery AB, Debs RJ, Luce JM, Corkery KJ, Turner J Hopewell PC. Aerosolized pentamidine as second line therapy in patients with AIDS with Pneumocystis carinii pneumonia. Chest 1989; 95: 747-750. [CrossRef]
- Conte JE, Chernoff D, Feigal DW, Joseph P, McDonald C, Golden JA. Intravenous or inhaled pentamidine isethionate for treating Pneumocystis carinii pneumonia in AIDS: a randomized trial. Ann Intern Med 1990; 113: 203-209. [CrossRef]
- Soo Hoo GW, Mohsenifar Z, Meyer RD. Inhaled or intravenous pentamidine therapy for Pneumocystis carinii pneumonia in AIDS: a randomized trial. Ann Intern Med 1990; 113: 195-202. [CrossRef]
- Hirschel B, Lazzarin A, Chopard P, Opravil M, Furer HJ, Rutiman S, et al. A controlled study of inhaled pentamidine for primary prevention of Pneumocystis carinii pneumonia. N Engl J Med 1991; 324: 1079-1083. [CrossRef]
- Leoung GS, Feigal DW, Montgomery AB, Wardlaw L, Adams M, Bush D, et al. Aerosolized pentamidine or prophylaxis against Pneumocystis carinii pneumonia. N Engl J Med. 1990; 323: 769-775. [CrossRef]
- Schneider MME, Hoepelman AIM, Schattenkerk JKME, Nielsen TL, van der Graaf Y, Frissen JPHJ, et al. A controlled trial of aerosolized pentamidine or trimethoprim-sulfamethoxazole as primary prophylaxis against Pneumocystis carinii pneumonia in patients with human immunodeficiency virus infection. N Engl J Med. 1992; 327: 1836-1841. [CrossRef]
- Hughes WT, Smith BL. Efficacy of diaminodiphenylsulfone and other drugs in murine Pneumocystis carinii pneumonitis. Antimicrob Agents Chemother. 1984; 26: 436-440. [CrossRef]
- 46. Leoung GS, Mills J, Hopewell PC, Hughes WT, Wofsy C. Dapsone-trimethoprim Pneumocystis carinii pneumonia in acquired immunodeficiency syndrome. Ann Intern Med 1986; 105: 45-48. [CrossRef]
- Mills J, Leoung G, Medina I, Hopewell PC, Hughes WT, Wofsy C. Dapsone treatment of Pneumocystis carinii pneumonia in the acquired immunodeficiency syndrome. Antimicrob Agents Chemother 1988; 32: 1057-1060. [CrossRef]
- Medina I, Mills J, Leoung G, Hopewell PC, Lee B, Modlin G, et al. Oral therapy for Pneumocystis carinii pneumonia in the acquired immunodeficiency syndrome. A controlled trial of trimethoprim-sulfamethoxazole versus trimethoprim-dapsone. N Engl J Med. 1990; 323: 776-782. [CrossRef]
- Hughes WT, Kennedy W, Dugdale M, Land MA, Stein DS, Weems JJ, et al. Prevention of Pneumocystis carinii pneumonia in AIDS patients with weekly dapsone. Lancet 1990; 336: 1066 [CrossRef]
- Lucas RC, Sandlund AM, Mijch A, Simpson JM. Primary dapsone chemoprophylaxis for Pneumocystis carinii pneumonia in immunocompromised patients infected with human immunodeficiency virus. Med J Aust. 1989; 151: 30-33.
- Metroka CF, Jacobus D, Lewis N. Successful chemoprophylaxis for Pneumocystis carinii with dapsone or Bactrim. Vth International Conference on AIDS. Montreal. June, 1989.
- Queener SF. DHFR and IMPDH: Enzymes explorable as drug targets in Pneumocystis. In, Walzer PD and Cushion MT. Pneumocystis Pneumonia. 3rd ed. Marcel Dekker, New York, 2005; 607-629.
- Hughes WT. From Wales to Pneumocystis and AIDS. Outskirts Press, Denver, 2015; pp. 347.
- Hughes WT, LaFon SW, Scott JD, Masur H. Adverse events associated with trimethoprim-sulfamethoxazole and atovaquone during the treatment of AIDS-related Pneumocystis carinii pneumonia. J Infect Dis. 1995; 171: 1295-1302. [CrossRef]
- Hughes WT, Dorenbaum A, Yogev R, Beauchamp B, Xu J, McNammara J, et al. A phase I safety and pharmacokinetics study of micronized atovaquone in human immunodeficiency virus (HIV)-infected infants and children. Antimicrob Agents Chemother. 1998; 42: 1315. [CrossRef]
- Madden RM, Pui CH, Hughes WT, Flynn PM, Leung W. Prophylaxis of Pneumocystis carinii with atovaquone in children with leukemia. Cancer. 2007; 15: 1654-1658. [CrossRef]
- El Sadr WM, Murphy RL, Yurik TM, Luskin-Hawk R, Cheung TW, Balfour HH, et al. Atovaquone compared with dapsone for the prevention of Pneumocystis carinii pneumonia in patients with HIV infection who cannot tolerate trimethoprim-sulfamethoxazole, sulfonamides or both. N Engl J Med. 1998; 339: 1889-1895. [CrossRef]
- Hughes WT, Oz HS. Successful prevention and prevention of babesiosis with atovaquone. J Infect Dis. 1995; 172: 1042-1046. [CrossRef]
- Hughes WT, Dankner WM, Yogev R, Huang S, Paul ME, Flores MA, et al. Comparison of atovaquone and azithromycin with trimethoprim-sulfamethoxazole for the prevention of serious bacterial infections in children with HIV. Clin Infect Dis. 2005; 40: 136-145. [CrossRef]
- Hughes WT, Sillos EM, LaFon S, Rogers M, Wooley JL, Davis C, et al. Effects of aerosolized synthetic surfactant, atovaquone, and the combination of these on murine Pneumocystis carinii pneumonia. J Infect Dis. 1998; 177:1046-1056. [CrossRef]
- Sheehan PM, Stokes DC, Yeh YY, Hughes WT. Surfactant phospholipids and lavage phospholipase A2 in experimental Pneumocystis carinii pneumonitis. Amer Rev Resp Dis 1986; 154: 315-322.
- Stokes DC, Hughes WT, Alderson PO, King RE, Garfinkel DJ. Lung mechanics, radiography, and 67Ga scintigraphy in experimental Pneumocystis carinii pneumonia. Br J Exper Pathol 1986; 67: 383-393.
- Kernbaum S, Masliah J, Alcindor LG, Bouton C, Christol D. Phospholipase activities of bronchoalveolar lavage fluid in rat Pneumocystis carinii pneumonia. Brit J Exp Pathol. 1983; 64: 75-80.
- Rice WR, Singleton FM, Linke MJ, Walzer PD. Regulation of surfactant phosphatidylcholine secretion from alveolar type cells during Pneumocystis carinii pneumonia in the rat. J Clin Invest. 1993; 92: 2778-2782. [CrossRef]
- Eijking EP, van Daal GJ, Tenbrinck R. Effect of surfactant replacement on Pneumocystis carinii replacement in rats. Intenswive Care Med. 1991; 17: 475-478. [CrossRef]
- Hoffman AGD, Lawrence MG, Ognibene FP, et al. Reduction of pulmonary surfactant in patients with human immunodeficiency virus infection and Pneumocystis carinii pneumonia. Chest. 1992; 102: 1730-1736. [CrossRef]
- Escamillia R, Precost MC, Hermant C, Caratero A, Cariven C, Krempf M. Surfactant analysis during Pneumocystis carinii pneumonia in HIV-infected patients. Chest 1992; 101: 1559-1562. [CrossRef]
- 68. Cushion MT, Walzer PD. Development of candidate of anti-Pneumocystis drugs: in vitro and in vivo approaches. Chapter 25 in, Walzer PD, Cushion MT. Pneumocystis Pneumonia, 3rd ed Marcel Dekker, New York, 2005.
- Schmatz DM, Romancheck MA, Pittarelli LA, Schwartz RE, Fromtling RA, Nollstadt KH, et al. Treatment of Pneumocystis carinii pneumonia with 1,3-β-glucan synthesis inhibitors. Proc Natl Acad Sci USA. 1990; 87: 5950-5954. [CrossRef]
- Cushion MT, Linke MJ, Ashbaugh A, Sesterhenn T, Collins MS, Lynch K, et al. Echinocandin treatment of pneumocystis pneumonia in rodent models depletes cysts leaving trophic burdens that cannot transmit the infection. Plos One. 2010; 5: e8524. [CrossRef]
- Lobo ML, Esteves F, de Sousa B, Cardoso F, Cushion MT, Antunes F. Therapeutic potential of caspofungin combined with trimethoprim-sulfamethoxazole for pneumocystis pneumonia: a pilot study in mice. Plos One. 2013; 8: e70619. [CrossRef]
- Aliouat EM, Dei-Cas, Gantois N, Pottier M, Pincon C, Hawser S, et al. In vitro and in vivo activity of iclaprim, a diaminopyrimidine compound and potential therapeutic alternative against Pneumocystis pneumonia. Euro J Clin Microbiol Infect Dis. 2018; 37: 409-415. [CrossRef]
- Gomes A, Ferraz R, Ficker L, Collins MS, Pudencio C, Cushion MT, et al. Chloroquine analogues as leads against Pneumocystis lung pathogens. Antimicrob Agents Chemother. 2018; 24: 62.e00983-18. [CrossRef]
- Huang H-B, Peng J-M, DU B. Echinocandins for Pneumocystis jirovecii pneumonia in non-HIV patients: a case report. Exper Therap Med. 2018; 16: 3227-3232.
- Armstrong D, Stebbing J, John L, Murrungi A, Bower M, Gazzard B, et al. A trial of caspofungin salvage treatment for PCP pneumonia. 2011; Thorax 66: 537-538. [CrossRef]
- Hughes WT, Smith-McCain BL. Effects of sulfonylurea on Pneumocystis carinii. J Infect Dis 1986; 153: 944-947. [CrossRef]
- Hughes WT, Smith BL, Jacobus DP. Successful treatment and prevention of murine Pneumocystis carinii pneumonia with 4,4’-sulfonylbisformanilide. Antimicrob Agents Chemother. 1986; 29: 509-510. [CrossRef]
- Hughes WT, Killmar J. Synergistic anti-Pneumocystis carinii effects of erythromycin and sulfamethoxazole. J Acquir Immune Defic Syndr 1991; 4: 532-537.
- Hughes WT. Macrolide-antifol synergism in anti-Pneumocystis carinii therapeutics. J Protozool. 1991; 38: 160S.
- Hughes WT, Jacobus DP, Canfield C, Killmar J. Anti-Pneumocystis carinii activity of PS-15, a new biguanide folate antagonist. Antimicrob Agents Chemother. 1993; 37: 1417-1449. [CrossRef]
- Hughes WT, Killmar JT, Oz HS. Relative potency of 10 drugs with anti-Pneumocystis carinii activity in an animal model. J Infect Dis. 1994; 170: 906-911. [CrossRef]
- Oz HS, Hughes WT. Novel anti-Pneumocystis carinii effects of the immunosuppressant mycophenolate mofetil in contrast to provocative effects of tacrolimus, sirolimus and dexamethasone. J Infect Dis. 1997; 175: 901-904. [CrossRef]
- Oz HS, Hughes WT, Rehg J. Efficacy of lasalocid against murine Pneumocystis carinii pneumonia. Antimicrob Agents Chemother 1997; 41: 191-192. [CrossRef]
- Wasserstein RL, Lazar NA. The ASA’s statement on P-values: context, process, and new purpose. Am Stat. 2016; 70: 129-133. [CrossRef]
- FDA Package insert. Bactrim™ (trimethoprim-sulfamethoxazole). www.iodine.om/drug/bactrim/fda-package-insert. 9/1/2018.
- Hughes WT. Postulates for the evaluation of adverse reactions to drugs. Clin Infect Dis. 1995; 20: 179-182. [CrossRef]
- Mon S, Sugimoto M. Pneumocystis jirovecii infection: an emerging threat to patients with rheumatoid arthritis. Rheumatology (Oxford) 2012; 51: 2120-2130. [CrossRef]

