

A is for Autophagy and Alzheimer's
Coad Thomas Dow 1, 2, †, * ![]() , Julia Szepieniec 3, †
, Julia Szepieniec 3, † ![]()
- McPherson Eye Research Institute, 9431 WIMR, 1111 Highland Ave., University of Wisconsin-Madison, WI 53705 USA
- Chippewa Valley Eye Clinic, 2715 Damon St., Eau Claire, WI 54701 USA
- Department of Biology and Chemistry, Upper Iowa University, 605 Washington St., Fayette, IA 52142 USA
† These authors contributed equally to this work.
* Correspondence: Coad Thomas Dow ![]()
Academic Editor: Michael Fossel
Received: October 1, 2018 | Accepted: November 21, 2018 | Published: November 26, 2018
OBM Geriatrics 2018, Volume 2, Issue 4 doi:10.21926/obm.geriatr.1804020
Recommended citation: Dow CT, Szepieniec J. A is for Autophagy and Alzheimer's. OBM Geriatrics 2018;2(4):020; doi:10.21926/obm.geriatr.1804020.
© 2018 by the authors. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
Abstract
Improved understanding of the underlying cellular dysfunction and resultant neuropathology of sporadic Alzheimer’s disease (AD) is needed to stem the anticipated public health crisis due to this increasingly common neurodegenerative disease. The four main risk factors for sporadic AD are age, female gender, genetic carriage of the APOE4 allele and type two diabetes mellitus (T2DM). Each of these four risk factors is associated with impaired and/or dysfunctional autophagy suggesting that perturbation of autophagy is a root contributor to AD. This article discusses normal cellular autophagy and how each of the named AD risk factors impacts autophagy; in addition currently existing interventions that favorably support the autophagic process are presented.
Keywords
Alzheimer’s; autophagy; Kallikrein 8; APOE4; pre-diabetes; insulin resistance; T2DM; telomerase; mTOR; caloric restriction
1. Introduction
Alzheimer’s disease (AD) is the most common cause of dementia having a worldwide prevalence of about 25 million in 2010; and according to the World Health Organization, that number is expected to double by 2030 due to increased life expectancy [1]. AD exacts a toll on families with affected love ones and on the health systems allocating resources to care for them. The burden of health care costs for patients with dementia in the last five years of life (287,038) far outweigh those of heart disease (175,136) and cancer (173,383) [2]. Unfolding understanding of the pathology of AD and the long prodromal period that precedes clinical dementia has yet to result in effective therapies. This article discusses how disruption of the autophagic process is a unifying feature of the four main risk factors for late-onset AD. Those main risk factors are: age, female gender, genetic APOE4 carriage, and insulin resistance.
Derived from Greek auto (oneself) and phagy (eat), autophagy refers to a common and necessary lysosomal degradation pathway of cellular “housekeeping.” Though this process has been studied for several decades, its broad role in biology has more recently increased understanding of molecular cellular processes; this includes recycling normal cellular molecules, protein quality control, pathogen elimination, regulation of immunity and inflammation, metabolism, and cell survival [3]. For his discovery of the mechanisms of autophagy, Yoshinori Ohsumi received the 2016 Nobel Prize in medicine.
The pathological proteins of AD are cerebral extracellular amyloid-β aggregation and intracellular tau tangles [4]. This pathology may be present decades before the onset of dementia [5,6]. Autophagy mediated intracellular removal of metabolic debris is impaired in the brains of AD patients [7]. The ubiquitin proteasome system (UPS) and autophagy are complementary mechanisms that accomplish important cellular housekeeping functions: removal of protein aggregates, turnover of organelles, and elimination of intracellular pathogens [8,9]. Ubiquitin-tagged macromolecules - “cargo” - are delivered to the lysosome by various methods: endocytosis for extracellular elements [10], autophagy for intracellular cargo [11], and xenophagy for pathogens [12]. These various cellular processes, employing “taking out the trash” functions, are commonly referred to as autophagy.
The remainder of this article will discuss how the four main risk factors of late-onset AD: age [13,14,15], female gender [1,16,17,18,19], APOE4 carriage [20,21,22], and insulin resistance [23,24,25] are associated with impaired and/or dysfunctional autophagy. Moreover, interventions that boost autophagy will be discussed. No human, animal, or plant subjects were a part of this article.
2. Age and Autophagy
The primary risk factor of AD is increasing age. The incidence of AD doubles every five years after the age of sixty-five [26,27]. Characterized by a broad progressive decline in physiologic function, aging exacts a marked toll on the immune system. Aging causes an inadequate initiation and resolution of the immune response; this is frequently termed immunosenescence which coexists with a persistent low-grade inflammatory state – inflammaging [28]. These chronic, age-driven, altered states have been linked to metabolic syndrome, cancer, atherosclerosis, and neurodegenerative disease including AD [29]. Microglia are the innate immune cells of the central nervous system performing important physiological functions. Conversely, malfunction of microglia is implicated in several pathological states [30,31]. On a cellular level, the aged microglia enter a state of senescence wherein it adopts a senescent phenotype that is found in the aged and diseased brain [32]; and when responding to CNS damage, by way of dysregulated autophagy, have limited capacity to clear aggregated protein [33,34]. The senescent state is due to telomere attrition. Telomeres are highly conserved repetitive DNA sequences that undergo shortening with each cell division (reflective of cellular replicative history) [35,36]. Accelerated telomere shortening is associated with AD [37]. Telomere length in T-cells of AD patients is shown to correlate with disease state suggesting immune alterations in AD pathogenesis [38]. While the connection between telomere attrition and autophagy in AD is nascent, the telomere attrition/autophagy association is quite well established in idiopathic pulmonary fibrosis (IPF), dyskeratosis congenita, and chronic obstructive pulmonary disease (COPD) [39,40].
Factors that accompany aging can confound the relative assignment of autophagic dysfunction to AD; e.g. age-related vascular disease and infection [41,42]. Contrasting with early thoughts whereby AD was mostly regarded as a singular disease, it is now well established that vascular insufficiency also plays a real, yet variable role in AD. This is not limited to circulation itself but also to age-related changes in the blood brain barrier (BBB) [43]. Additionally, head injury is associated with AD [44]; and, trauma-induced vascular changes are linked to AD pathology [45].
Moreover, infectious burden is felt to play a role in AD [46]. In an age-dependent manner, most of the population harbours latent herpes virus infections acquired during their lifetimes. While infection rates in neonates are very low, in the case of herpes simplex virus type 1 (HSV1), by age 70 years 80–90% of the population is seropositive. The virus is present in the brain [47] of many elderly individuals and AD patients; it was proposed that sporadic reactivation of latent HSV1 in the brain, particularly in APOE 4 carriers, might confer an increased risk of later developing AD [47]. Strikingly, when patients were aggressively treated with anti-herpetic medications at the time of their initial infection, the relative risk of senile dementia (both vascular dementia and AD) was reduced by a factor of 10 [48]. Similarly, age-related infection by cytomegalovirus (CMV) plays a role in diverting the immune capacity [49,50]. CMV is associated with an age-related prevalence ranging from 40% in twenty-year-olds to over 90% in eighty-year-olds [51]. It has been suggested that subclinical viral infection, exacting an exhaustive toll on immune function and attendant impaired autophagy, can lead to AD [52].
3. Gender and Autophagy
Women have a greater prevalence for AD than men accounting for two-thirds of all patients [53]. This is only partially explained by increased premature mortality of men (starting at 45 years); women aged 65 and older are twice as likely to develop AD compared to age-matched men [54]. Women have faster progression of mild cognitive impairment (MCI) [55]; and women have, after 80 years, a higher transition rate from MCI to AD [56]. Moreover, women display a greater severity in their clinical dementia [57,58,59]. The use of estrogen replacement for treatment of AD is controversial. However, there are observations that suggest that estradiol may decrease aggregates of amyloid by stimulating autophagy [60,61]. Estrogen, as a therapeutic agent for neurodegenerative disease, has been investigated over the last decade. E2 (17β-estradiol) has been shown to be neuroprotective in a variety of neurodegenerative disorders, including Parkinson's disease and Alzheimer's disease [62,63]. Nascent evidence supports the concept that activation of autophagy by E2 is protective in neurodegenerative diseases and acts by enhancing the removal of toxic protein aggregates [64]. Similarly, in animal studies sex differences in disease-related cognitive impairments and pathology burden are detected in various transgenic mouse models of AD. Female mice are more severely affected by cognitive deficits [65,66,67] and amyloid plaque pathology than males [68,69,70,71]; and estrogen is implicated in the sex differences in the AD mouse models [72]. Estrogen loss via ovary removal increases amyloid burden in female mice [73] while estrogen replacement reduces amyloid burden [74].
Recent studies searching for underlying female-specific risk for AD-related pathology identify kallikrein-8 (KLK8) protease as such a factor. Estradiol, but not testosterone, induces KLK8 synthesis in neuronal and microglial cells. The significant sex-specific difference in AD may be mediated by estrogen-induced overproduction of KLK8 in prodromal AD. Inhibition of KLK8 enhances autophagy. A higher level of KLK8 is detected in brains of female Alzheimer’s disease patients at early disease stages compared with male counterparts. These studies conclude by suggesting that KLK8 overexpression and accompanying impaired autophagy in females is central to the preferential prevalence of AD in females [75].
4. APOE and Autophagy
Unlike early-onset familial AD, which accounts for less than one percent of AD cases and typically affects before the age of 65, the vast majority of AD cases occur at over 65 years of age and are most commonly referred to as late-onset AD (LOAD) [18]. LOAD is thought to have multiple genetic and environmental risk factors [76]. The inheritance of both APOE4 alleles (E4/E4) is the single greatest genetic risk for the development of AD [77,78,79]. The human APOE gene exists as three different alleles: E2, E3 and E4. They have a respective frequency of 8.4%, 77.9% and 13.7% (Figure 1) [20]. A single E4 allele increases AD risk by 2-3 folds. Having both alleles increases AD risk by about 12-fold [80,81]. While APOE4 is strongly associated with AD, this allele is only found in half of LOAD cases [77,78,79,80,81,82,83]. APOE4 is associated with impaired memory and rapidity of memory decline as well as increased risk of progression from MCI (mild cognitive impairment) to AD dementia [84,85].
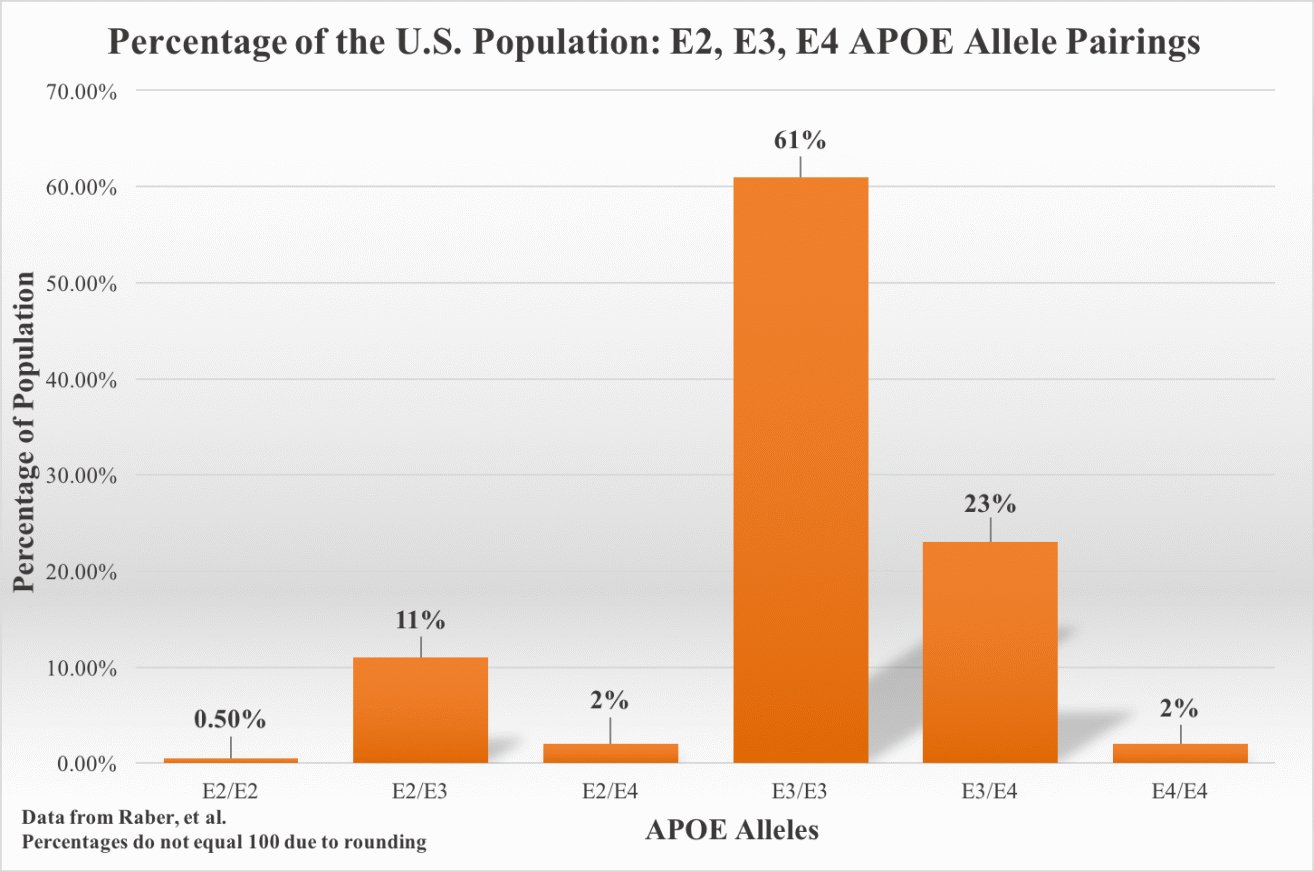
Figure 1 Percentage of the U.S. Population: E2, E3, E4 APOE Allele Pairings [86].
Impaired autophagy is known to be both an early and a persistent finding of AD pathogenesis. APOE is abundantly produced in the brain and serves as the main lipid transport, and its messenger RNA is found in the regions of the brain that demonstrate AD pathology: the hippocampus, cerebral cortex, cerebellum, and medulla [87]. Autophagy reduces protein aggregation and studies show APOE4 competes with a transcription factor that controls autophagic gene expression. There is decreased autophagic activity in AD carriers of APOE4 relative to those with allele E2 and E3 which correlates with increased aggregated amyloid and tau proteins in the E4 carriers [88,89].
5. Insulin Resistance and Autophagy
Diabetes mellitus is a chronic metabolic disease characterized by hyperglycemia resulting from deficient insulin production and/or deficient insulin action. The International Diabetes Federation reports that type 2 diabetes (T2DM), the so-called noninsulin-dependent diabetes, accounts for approximately 90% of all cases of diabetes and affects nearly 415 million people worldwide [90]. In contrast to type 1 diabetes (T1DM), where little or no insulin is produced by the pancreas due to an autoimmune-mediated destruction of β -cells, T2DM arises as a result of hyperinsulinemia accompanied by impaired glucose tolerance, which is believed to be a result of compensatory insulin signaling impairment in peripheral tissues [91,92]. According to the CDC, more than 1 in 3 Americans have prediabetes and the majority are unaware of it [93]. Approximately 70% of individuals with prediabetes develop T2DM [94] and an estimated 35% of Americans over the age of 20 are pre-diabetic, while approximately half of the individuals over the age of 65 are affected by diabetes [95]. Peripheral insulin resistance is associated with compensatory insulin hypersecretion and decreased hepatic insulin clearance, rendering to a pathologic cycle of both high blood glucose and high insulin levels [96,97]. Autophagy plays an important role in pancreatic cell dysfunction of T2DM and insulin resistance [98,99].
Though the brain holds about 2% of total body mass, it consumes about 20% of total body energy; this disproportionate energy demand is due to constant brain action even when at rest [99]. Brain levels of insulin and insulin receptors (IR) are lower in AD, and insulin signaling impairments have been documented in both AD post-mortem analysis and in animal models of AD. Accumulating evidence suggests that insulin resistance also develops in AD brains [100,101,102,103]. In the light of the epidemiological data supporting the link between T2D and AD, the increasing incidence of AD is likely a consequence not only of ageing alone, but of the diabetes epidemic itself [28]. It has been hypothesized that glucose transportation abnormalities may be related to insulin resistance and intracellular metabolic alterations to mitochondrial dysfunction observed in AD patients [104]. These changes happen decades prior to clinical symptoms. Cross-sectional positron emission tomography (PET) studies find that cognitively normal carriers of the APOE4 allele have abnormally low measurements of the cerebral metabolic rate for glucose (CMRgl) in the same regions as patients with Alzheimer’s dementia [105].
The impaired signaling of insulin and insulin-like growth factor (IGF) that accompanies diabetes also disrupts autophagy. This imparts the core pathological features of AD: amyloid plaque and a modification of the tau protein producing the neurofibrillary tangle [23].
6. Discussion: Upregulating Autophagy
While there is no avoiding “the ravages of time,” there is a rationale to consider strategies offering molecular support that increases autophagy in the aging brain. The mammalian target of rapamycin (mTOR) signaling pathway is a well described controller of autophagy. Neurons are small, polarized, and post-mitotic making them sensitive to the accumulation of aggregated and damaged cellular proteins. Neurons are thus dependent upon autophagy for survival [106].
mTOR responds to cellular levels of available glucose. Under normal conditions where there is no cell starvation, mTORC1 suppresses autophagy [107]. Under starvation conditions or rapamycin treatment, mTOR activity is inhibited, leading to autophagy initiation [108].
Rapamycin and rapalogs are protein kinases that inhibit mTOR [109]. Rapamycin has been shown to slow age-dependent changes in animal models [110]. mTOR signaling pathway is a master regulator of cell growth and metabolism [111]. Mounting evidence suggests that AD as well as Parkinson’s, Huntington’s, and spinocerebellar ataxia may all be related to mTOR protein synthesis and impaired autophagy [112]. Another autophagy inhibitor that acts by inhibiting mTOR is the antidepressant indatraline [113].
As described earlier, age-related telomere shortening is a recognized factor in the pathogenesis of AD [38]. Telomere length in T-cells of AD patients is shown to correlate with disease state suggesting immune alterations in AD pathogenesis [39]. A cellular ribonucleoprotein enzyme complex, telomerase, “rescues” the cell from the senescent state (and senescent phenotype) by adding telomeres [114,115]. In a small study an oral telomerase activator has been used to benefit early age-related macular degeneration (AMD) [116] as a health supplement and may find its way into treatment of AD [117]. Attempts are also underway to develop a method to upregulate telomerase via an injectable telomerase activator [118].
The use of estrogen as a therapeutic agent for neurodegenerative diseases has been investigated over the last decade. E2 (17β-estradiol) has been shown to be beneficial in a number of neurodegenerative disorders, such as stroke, Parkinson's disease, and Alzheimer's disease [62,63]. Estrogen may also exert undesirable stimulatory effects on the breast and uterus, potentially increasing the risk of developing breast and uterine cancers or stroke [62]. These limitations have prompted development and trials of selective estrogen receptor modulators (SERMs), a treatment that mimics the beneficial yet minimizes the adverse effects of estrogen. One widely used SERM drug is raloxifene (Ral). In vitro and in vivo studies have shown Ral demonstrates neuroprotection in animal models of neurodegenerative disease [119]. As described earlier, the significant sex-specific differences in AD are related to estrogen-induced overproduction of KLK8 in prodromal AD. KLK8 is manifest in AD affected areas: the hippocampus, frontal cortex and cerebellum [75]. Inhibition of KLK8 enhances autophagy and, in animal models, reduces amyloid and tau pathology [120].
We cannot change our genetics yet “knowledge is power” [121]. An individual’s knowing that they carry one or both alleles of the APOE4 gene may make them adopt lifestyle changes to their long term cognitive benefit. APOE4 regulates pathways that link aging and AD; APOE4 expression reduces the activity of Sirtuin1 (SirT1), a protein that suppresses AD related pathology. Most of the cellular abnormalities associated with APOE4 and AD could be prevented by increasing SirT1 [122]. While we cannot change our genetics there is reason to believe that we can influence our epigenetics, via telomerase activation [123]; not only has this been proposed, but human FDA trials are planned [124]. Caloric restriction (30-60% below ad libitum in animal studies) can increase lifespan by 20-50% [125] and does so by activating SirT1 [126,127]. There is active research looking for caloric restriction mimetics [128]. Knowledge of APOE4 carriage would allow individuals to fully explore the lifestyle changes to mitigate AD; those include sleep, diet and cortisol regulations [129]. Moreover, regular exercise has been shown to benefit AD risk [130] as well as the symptoms and pathology of AD [131].
AD is occasionally termed “type 3 diabetes” due to insulin deficiency and/or insulin resistance that is specific to the brain. As insulin is the primary treatment for diabetes, it is understandable that small clinical trials in patients with mild cognitive impairment/AD, nasal application of insulin or long-lasting insulin analogues, showed improvements in memory tasks, cerebrospinal fluid biomarkers, and in a fluorodeoxyglucose positron (PET) emission tomographic study [132]. However, in further studies, responses varied by APOE genotype and gender [133].
What is available today then for an elderly, female, APOE4 positive, and insulin resistant individual? Certainly, there are lifestyle recommendations regarding diet, sleep, exercise and stress reduction that may deter and defer the cognitive changes of AD. For tomorrow perhaps there will be telomerase activators, mTOR inhibitors, caloric restriction mimetics, KLK8 inhibitors, SirT1 activators, intranasal insulin, and antiviral treatments that may benefit those who are risk of developing AD dementia.
Author Contributions
CTD chose the topic for this article. CTD and JS contributed equally to the research and writing of this article.
Competing Interests
The authors have declared that no competing interests exist.
References
- World Health Organization, Alzheimer’s Disease International. Dementia: a public health priority [Internet]. United Kingdom: World Health Organization; 2012 [cited 2018 July 24]. Available from: http://www.who.int/mental_health/publications/dementia_report_2012/en/.
- Kelley AS, McGarry K, Gorges R, Skinner JS. The burden of health care costs for patients with dementia in the last 5 years of life. Ann Intern Med. 2015; 163: 729-736. [CrossRef]
- Levine B, Klionsky DJ. Autophagy wins the 2016 Nobel Prize in Physiology or Medicine: Breakthroughs in baker's yeast fuel advances in biomedical research. Proc Natl Acad Sci USA. 2017; 114: 201-205. [CrossRef]
- Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012; 367: 795-804. [CrossRef]
- Jack CR Jr., Holtzman DM. Biomarker modeling of Alzheimer’s disease. Neuron. 2013; 80: 1347-1358. [CrossRef]
- Sutphen CL, Jasielec MS, Shah AR, Macy EM, Xiong C, Vlassenko AG, et al. Longitudinal cerebrospinal fluid biomarker changes in preclinical Alzheimer disease during middle age. JAMA Neurol. 2015; 72: 1029-1042. [CrossRef]
- Ghavami S, Shojaei S, Yeganeh B, Ande SR, Jangamreddy JR, Mehrpour M, et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol. 2014; 112: 24-49. [CrossRef]
- Correia SC, Resende R, Moreira PI, Pereira CM. Alzheimer’s disease related misfolded proteins and dysfunctional organelles on autophagy menu. DNA Cell Biol. 2015; 34: 261-273. [CrossRef]
- Magraoui FE, Reidick C, Meyer HE, Platta HW. Autophagy-related deubiquitinating enzymes involved in health and disease. Cells. 2015; 4: 596-621. [CrossRef]
- Platta HW, Stenmark H. Endocytosis and signaling. Curr Opin Cell Biol. 2011; 23: 393-403. [CrossRef]
- Ryter SW, Cloonan SM, Choi AM. Autophagy: A critical regulator of cellular metabolism and homeostasis. Mol Cells. 2013; 36: 7-16. [CrossRef]
- Sorbara MT, Girardin SE. Emerging themes in bacterial autophagy. Curr Opin Microbiol. 2015; 23: 163-170. doi: 10.1016/j.mib.2014.11.020. [CrossRef]
- Brookmeyer R, Gray S, Kawas C. Projections of Alzheimer’s disease in the United States and the public health impact of delaying disease onset. Am J Public Health. 1998; 88: 1337-1342. [CrossRef]
- Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement. 2007; 3: 186-191. [CrossRef]
- Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology. 2013; 80: 1778-1783. [CrossRef]
- Payami H, Montee KR, Kaye JA, Bird TD, Yu CE, Wijsman EM, et al. Alzheimer’s disease, apolipoprotein E4, and gender. JAMA. 1994; 271: 1316-1317. [CrossRef]
- Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: a meta-analysis. JAMA. 1997; 278: 1349-1356. [CrossRef]
- Mortensen EL, Hogh P. A gender difference in the association between APOE genotype and age-related cognitive decline. Neurology. 2001; 57: 89-95. [CrossRef]
- Damoiseaux JS, Seeley WW, Zhou J, Shirer WR, Coppola G, Karydas A, et al. Gender modulates the APOE epsilon4 effect in healthy older adults: convergent evidence from functional brain connectivity and spinal fluid tau levels. J Neurosci. 2012; 32: 8254-8262. [CrossRef]
- Mayeux R, Stern Y, Ottman R, Tatemichi TK, Tang MX, Maestre G, et al. The apolipoprotein epsilon 4 allele in patients with Alzheimer’s disease. Ann Neurol. 1993; 34: 752-754. [CrossRef]
- Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E: structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J Lipid Res. 2009; 50(Suppl): S183-S188. [CrossRef]
- Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013; 9: 106-118. [CrossRef]
- de la Monte SM, Wands JR. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J Diabetes Sci Technol. 2008; 2: 1101-1113. [CrossRef]
- Ahmed S, Mahmood Z, Zahid S. Linking insulin with Alzheimer’s disease: emergence as type III diabetes. Neurol Sci. 2015; 36: 1763-1769. [CrossRef]
- Ribe EM, Lovestone S. Insulin signalling in Alzheimer’s disease and diabetes: from epidemiology to molecular links. J Intern Med. 2016; 280: 430-442. [CrossRef]
- Jorm AF, Korten AE, Henderson AS. The prevalence of dementia: a quantitative integration of the literature. Acta Psychiatr Scand. 1987; 76: 465-479. [CrossRef]
- Ritchie K, Kildea D, Robine JM. The relationship between age and the prevalence of senile dementia: a meta-analysis of recent data. Int J Epidemiol. 1992; 21: 763-769. [CrossRef]
- Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F, et al. Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev. 2007; 128: 92-105. [CrossRef]
- Wong WT. Microglial aging in the healthy CNS: phenotypes, drivers, and rejuvenation. Front Cell Neurosci. 2013; 7: 22. [CrossRef]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005; 308: 1314-1318. [CrossRef]
- Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011; 91: 461-553. [CrossRef]
- Deleidi M, Jäggle M, Rubino G. Immune aging, dysmetabolism, and inflammation in neurological diseases. Front Neurosci. 2015; 9: 172. [CrossRef]
- Plaza-Zabala A, Sierra-Torre V, Sierra A. Autophagy and microglia: Novel partners in neurodegeneration and aging. Int J Mol Sci. 2017; 18: 598. [CrossRef]
- Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci. 2008; 28: 8354-8360. [CrossRef]
- Flanary BE, Streit WJ. Telomeres shorten with age in rat cerebellum and cortex in vivo. J Anti Aging Med. 2003; 6: 299-308. [CrossRef]
- Satyanarayana A, Greenberg RA, Schaetzlein S, Buer J, Masutomi K, Hahn WC, et al. Mitogen stimulation cooperates with telomere shortening to activate DNA damage responses and senescence signaling. Mol Cell Biol. 2004; 24: 5459-5474. [CrossRef]
- Liu M, Huo YR, Wang J, Wang C, Liu S, Liu S, et al. Telomere shortening in Alzheimer's disease patients. Ann Clin Lab Sci. 2016; 46: 260-265.
- Panossian LA, Porter VR, Valenzuela HF, Zhu X, Reback E, Masterman D, et al. Telomere shortening in T cells correlates with Alzheimer’s disease status. Neurobiol Aging. 2003; 24: 77-84. [CrossRef]
- Kuwano K, Araya J, Hara H, Minagawa S, Takasaka N, Ito S, et al. Cellular senescence and autophagy in the pathogenesis of chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF). Respir Investig. 2016; 54: 397-406. [CrossRef]
- Bertuch AA. The molecular genetics of the telomere biology disorders. RNA Biol. 2016; 13: 696-706. [CrossRef]
- Cacabelos R, Fernandez-Novoa L, Lombardi V, Corzo L, Pichel V, Kubota Y. Cerebrovascular risk factors in Alzheimer’s disease: brain hemodynamics and pharmacogenomic implications. Neurol Res. 2003; 25: 567-580. [CrossRef]
- Gorelick PB. Risk factors for vascular dementia and Alzheimer disease. Stroke. 2004; 35: 2620-2622. [CrossRef]
- Cai Z, Qiao PF, Wan CQ, Cai M, Zhou NK, Li Q. Role of blood-brain barrier in Alzheimer's disease. J Alzheimers Dis. 2018; 63: 1223-1234. [CrossRef]
- van Duijn CM, Tanja TA, Haaxma R, Schulte W, Saan RJ, Lameris AJ, et al. Head trauma and the risk of Alzheimer's disease. Am J Epidemiol. 1992; 135: 775-782. [CrossRef]
- Ramos-Cejudo J, Wisniewski T, Marmar C, Zetterberg H, Blennow K, de Leon MJ, et al. Traumatic brain injury and Alzheimer's disease: The Cerebrovascular Link. EBioMedicine. 2018; 28: 21-30. [CrossRef]
- Bu XL, Yao XQ, Jiao SS, Zeng F, Liu YH, Xiang Y, et al. A study on the association between infectious burden and Alzheimer's disease. Eur J Neurol. 2015; 22: 1519-25. [CrossRef]
- Itzhaki RF, Lin WR, Shang D, Wilcock GK, Faragher B, Jamieson GA. Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet. 1997; 349: 241-244. [CrossRef]
- Tzeng N-S, Chung CH, Lin FH, Chiang CP, Yeh CB, Huang SY, et al. Antiherpetic medications and reduced risk of dementia in patients with herpes simplex virus infections-A nationwide population based cohort study in Taiwan. Neurotherapeutics. 2018; 15: 417-429. [CrossRef]
- Westman G, Berglund D, Widén J, Ingelsson M, Korsgren O, Lannfelt L, et al. Increased inflammatory response in cytomegalovirus seropositive patients with Alzheimer’s disease. Plos One. 2014; 9: e96779. [CrossRef]
- Lurain NS, Hanson BA, Martinson J, Leurgans SE, Landay AL, Bennett DA, et al. Virological and immunological characteristics of human cytomegalovirus infection associated with Alzheimer disease. J Infect Dis. 2013; 208: 564-572. [CrossRef]
- Badami KG, McQuilkan-Bickerstaffe S, Wells JE, Parata M. Cytomegalovirus seroprevalence and ‘cytomegalovirus‐safe’ seropositive blood donors. Epidemiol Infect. 2009; 137: 1776-1780. [CrossRef]
- Dow T. CMV Driven immunosenescence and Alzheimer’s disease. J Neuroinfect Dis. 2015; 6: 4.
- Brookmeyer R, Evans DA, Hebert L, Langa KM, Heeringa SG, Plassman BL, et al. National estimates of the prevalence of Alzheimer’s disease in the United States. Alzheimers Dement. 2011; 7: 61-73. [CrossRef]
- Thies W, Bleiler L. 2013 Alzheimer’s disease facts and figures. Alzheimers Dement. 2013; 9: 208-245. [CrossRef]
- Lin KA, Choudhury KR, Rathakrishnan BG, Marks DM, Petrella JR, Doraiswamy PM. Marked gender differences in progression of mild cognitive impairment over 8 years. Alzheimers Dement. 2015; 1: 103-110. [CrossRef]
- Roberts RO, Geda YE, Knopman DS, Cha RH, Pankratz VS, Boeve BF, et al. The incidence of MCI differs by subtype and is higher in men: the Mayo Clinic Study of Aging. Neurology. 2012; 78: 342-351. [CrossRef]
- Irvine K, Laws KR, Gale TM, Kondel TK. Greater cognitive deterioration in women than men with Alzheimer’s disease: a meta analysis. J Clin Exp Neuropsychol. 2012; 34: 989-998. [CrossRef]
- Mielke MM, Vemuri P, Rocca WA. Clinical epidemiology of Alzheimer’s disease: assessing sex and gender differences. Clin Epidemiol. 2014; 6: 37-48. [CrossRef]
- Pike CJ. Sex and the development of Alzheimer’s disease. J Neurosci Res. 2017; 95: 671-680. [CrossRef]
- Pike CJ, Carroll JC, Rosario ER, Barron AM. Protective actions of sex steroid hormones in Alzheimer’s disease. Front Neuroendocrinol. 2009; 30: 239-258. [CrossRef]
- Simpkins JW, Yi KD, Yang SH, Dykens JA. Mitochondrial mechanisms of estrogen neuroprotection. Biochim Biophys Acta. 2010; 1800: 1113-1120. [CrossRef]
- Brann DK, Wakade C, Mahesh VB, Khan MM. Neurotrophic and neuroprotective actions of estrogen: basic mechanisms and clinical implications. Steroids. 2007; 72: 381-405. [CrossRef]
- Henderson VW. Cognitive changes after menopause: influence of estrogen. Clin Obstet Gynecol. 2008; 51: 618-626. [CrossRef]
- Cheung ZH, Ip NY. Autophagy deregulation in neurodegenerative diseases - recent advances and future perspectives. J Neurochem. 2011; 118: 317-325. [CrossRef]
- Clinton LK, Billings LM, Green KN, Caccamo A, Ngo J, Oddo S, et al. Age-dependent sexual dimorphism in cognition and stress response in the 3xTg-AD mice. Neurobiol Dis. 2007; 28: 76-82. [CrossRef]
- King DL, Arendash GW, Crawford F, Sterk T, Menendez J, Mullan MJ. Progressive and gender-dependent cognitive impairment in the APP(SW) transgenic mouse model for Alzheimer’s disease. Behav Brain Res. 1999; 103: 145-162. [CrossRef]
- Pistell PJ, Zhu M, Ingram DK. Acquisition of conditioned taste aversion is impaired in the amyloid precursor protein/presenilin 1 mouse model of Alzheimer’s disease. Neuroscience. 2008; 152: 594-600. [CrossRef]
- Callahan MJ, Lipinski WJ, Bian F, Durham RA, Pack A, Walker LC. Augmented senile plaque load in aged female beta-amyloid precursor protein-transgenic mice. Am J Pathol. 2001; 158: 1173-1177. [CrossRef]
- Hirata-Fukae C, Li HF, Hoe HS, Gray AJ, Minami SS, Hamada K, et al. Females exhibit more extensive amyloid, but not tau, pathology in an Alzheimer transgenic model. Brain Res. 2008; 1216: 92-103. [CrossRef]
- Kulnane LS, Lamb BT. Neuropathological characterization of mutant amyloid precursor protein yeast artificial chromosome transgenic mice. Neurobiol Dis. 2001; 8: 982-992. [CrossRef]
- Wang J, Tanila H, Puolivali J, Kadish I, van Groen T. Gender differences in the amount and deposition of amyloidbeta in APPswe and PS1 double transgenic mice. Neurobiol Dis. 2003; 14: 318-327. [CrossRef]
- Yue X, Lu M, Lancaster T, Cao P, Honda SI, Staufenbiel M, et al. Brain estrogen deficiency accelerates Ab plaque formation in an Alzheimer’s disease animal model. Proc Natl Acad Sci USA. 2005; 102: 19198-19203. [CrossRef]
- Zheng H, Xu H, Uljon S, Gross R, Hardy K, Gaynor J, et al. Modulation of A peptides by estrogen in mouse models. J Neurochem. 2001; 80: 191-196. [CrossRef]
- Huang J, Guan H, Booze RM, Eckman CB, Hersh LB. Estrogen regulates neprilysin activity in rat brain. Neurosci Lett. 2004; 367: 85-87. [CrossRef]
- Keyvani K, Munster Y, Kurapati NK, Rubach S, Schonborn A, Kocakavuk E, et al. Higher levels of kallikrein-8 in female brain may increase the risk for Alzheimer’s disease. Brain Pathol. 2018. [CrossRef]
- Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, et al. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science. 2010; 330: 1774. [CrossRef]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GC, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993; 261: 921-923. [CrossRef]
- Bu G. Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. 2009; 10: 333-344. [CrossRef]
- Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012; 148: 1204-1222. [CrossRef]
- Mahley RW, Rall SC Jr. Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet. 2000; 1: 507-537. [CrossRef]
- Verghese PB, Castellano JM, Holtzman DM. Apolipoprotein E in Alzheimer’s disease and other neurological disorders. Lancet Neurol. 2011; 10: 241-252. [CrossRef]
- Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA. 1993; 90: 1977-1981. [CrossRef]
- Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer's disease. Neurology. 1993; 43: 1467-1472. [CrossRef]
- Ramakers IH, Visser PJ, Aalten P, Bekers O, Sleegers K, van Broekhoven CL, et al. The association between APOE genotype and memory dysfunction in subjects with mild cognitive impairment is related to age and Alzheimer pathology. Dement Geriatr Cogn Disord. 2008; 26: 101-108. [CrossRef]
- Dik MG, Jonker C, Bouter LM, Geerlings MI, van Kamp GJ, Deeg DJH. APOE-ε4 is associated with memory decline in cognitively impaired elderly. Neurology. 2000; 54: 1492-1497. [CrossRef]
- Raber J, Huang Y, Ashford JW. ApoE genotype accounts for the vast majority of AD risk and AD pathology. Neurobiol Aging. 2004; 25: 641-650. [CrossRef]
- Huang Y, Mahley RW. Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol Dis. 2000; 72(Pt A): 3-12.
- Christensen DZ, Schneider-Axmann T, Lucassen PJ, Bayer TA, Wirths O. Accumulation of intraneuronal Abeta correlates with ApoE4 genotype. Acta Neuropathol. 2010; 119: 555-566. [CrossRef]
- Aboud O, Parcon PA, DeWall KM, Liu L, Mrak RE, Griffin WS. Aging, Alzheimer’s, and APOE genotype influence the expression and neuronal distribution patterns of microtubule motor protein dynactin- P50. Front Cell Neurosci. 2015; 9: 103. [CrossRef]
- International Diabetes Federation. IDF Diabetes Atlas. 7th ed. International Diabetes Federation; 2015. Available from: http://diabetesatlas.org/IDF_Diabetes_Atlas_8e_interactive_EN/.
- Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci USA. 2003; 100: 4162-4167. [CrossRef]
- Abdul-Hay SO, Kang D, McBride M, Li L, Zhao J, Leissring MA. Deletion of insulin-degrading enzyme elicits antipodal, age-dependent effects on glucose and insulin tolerance. Plos One. 2011; 6: e20818. [CrossRef]
- CDC. Prediabetes: Your Chance to Prevent Type 2 Diabetes [Internet]. Atlanta (GA): Centers for Disease Control and Prevention; 2018 [cited 2018 July 17]. Available from: https://www.cdc.gov/diabetes/basics/prediabetes.html.
- Wang D, Zhao N, Zhu Z. Recombinant human growth hormone in treatment of diabetes: report of three cases and review of relative literature. Int J Clin Exp Med. 2015; 8: 8243-8248.
- Centers for Disease Control and Prevention. National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States [Internet]. Atlanta (GA): U.S. Department of Health and Human Services, Centers for Disease Control and Prevention; 2011 [cited 2018 July 17]. Available from: https://www.cdc.gov/diabetes/pubs/pdf/ndfs_2011.pdf.
- Del Prato S, Marchetti P. Beta and alpha-cell dysfunction in type 2 diabetes. Horm Metab Res. 2004; 36: 775-781. [CrossRef]
- Mayfield JA, White RD. Insulin therapy for type 2 diabetes: Rescue, augmentation and replacement of beta-cell function. Am Fam Physician. 2004; 70: 489-500.
- Yang JS, Lu CC, Kuo SC, Hsu YM, Tsai SC, Chen SY, et al. Autophagy and its link to type II diabetes mellitus. Biomedicine (Taipei). 2017; 7: 8. [CrossRef]
- Shetty PK, Galeffi F, Turner DA. Cellular links between neuronal activity and energy homeostasis. Front Pharmacol. 2012; 3: 43. [CrossRef]
- de la Monte SM. Insulin resistance and Alzheimer’s disease. BMB Rep. 2009; 42: 475-481. [CrossRef]
- Moloney AM, Griffin RJ, Timmons S, O’Connor R, Ravid R, O’Neill C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol Aging. 2010; 31: 224-243. [CrossRef]
- Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, et al. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease - is this type 3 diabetes? J Alzheimers Dis. 2005; 7: 63-80. [CrossRef]
- Craft S. Insulin resistance and Alzheimer’s disease pathogenesis: potential mechanisms and implications for treatment. Curr Alzheimer Res. 2007; 4: 147-152. [CrossRef]
- Chen Z, Zhong C. Decoding Alzheimer's disease from perturbed cerebral glucose metabolism: implications for diagnostic and therapeutic strategies. Prog Neurobiol. 2013; 108: 21-43. [CrossRef]
- Reiman EM, Caselli RJ, Chen K, Alexander GE, Bandy D, Frost J. Declining brain activity in cognitively normal apolipoprotein E epsilon 4 heterozygotes: A foundation for using positron emission tomography to efficiently test treatments to prevent Alzheimer's disease. Proc Natl Acad Sci USA. 2001; 98: 3334-3339. [CrossRef]
- Tooze SA, Schiavo G. Liaisons dangereuses: autophagy, neuronal survival and neurodegeneration. Curr Opin Neurobiol. 2008; 18: 504-515. [CrossRef]
- Wojcik S. Crosstalk between autophagy and proteasome protein degradation systems: possible implications for cancer therapy. Folia Histochem Cytobiol. 2013; 51: 249-264. [CrossRef]
- Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010; 22: 124-131. [CrossRef]
- Ballou LM, Lin RZ. Rapamycin and mTOR kinase inhibitors. J Chem Biol. 2008; 1: 27-36. [CrossRef]
- Wilkinson JE, Burmeister L, Brooks SV, Chan CC, Friedline S, Harrison DE, et al. Rapamycin slows aging in mice. Aging Cell. 2012; 11: 675-682. [CrossRef]
- Li J, Kim SG, Blenis J. Rapamycin: one drug, many effects. Cell Metab. 2014; 19: 373-379. [CrossRef]
- Bové J, Martínez-Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci. 2011; 12: 437-452. [CrossRef]
- Cho YS, Yen CN, Shim JS, Kang DH, Kang SW, Liu JO, et al. Antidepressant indatraline induces autophagy and inhibits restenosis via suppression of mTOR/S6 kinase signaling pathway. Sci Rep. 2016; 6: 34655. [CrossRef]
- Wong JM, Collins K. Telomere maintenance and disease. Lancet. 2003; 362: 983-988. [CrossRef]
- Blackburn EH, Greider CW, Szostak JW. Telomeres and telomerase: the path from maize, Tetrahymena and yeast to human cancer and aging. Nat Med. 2006; 12: 1133-1138. [CrossRef]
- Dow CT, Harley CB. Evaluation of an oral telomerase activator for early age-related macular degeneration - a pilot study. Clin Ophthalmol. 2016; 10: 243-249. [CrossRef]
- Harley CB, Liu W, Flom PL, Raffaele JM. A natural product telomerase activator as part of a health maintenance program: metabolic and cardiovascular response. Rejuvenation Res. 2013; 16: 386-395. [CrossRef]
- Fossel M. The telomerase revolution: The enzyme that holds the key to human aging and will soon lead to longer, healthier lives. 1st ed. Dallas, TX: BenBella Books, Inc.; 2015.
- Arevalo M, Santos-Galindo M, Lagunas N, Azcoitia I, Garcia-Segura L. Selective estrogen receptor modulators as brain therapeutic agents. J Mol Endocrinol. 2011; 46: R1-R9. [CrossRef]
- Herring A, Munster Y, Akkaya T, Moghaddam S, Deinsberger K, Meyer J, et al. Kallikrein-8 inhibition attenuates Alzheimer’s disease pathology in mice. Alzheimers Dement. 2016; 12: 1273-1287. [CrossRef]
- Scientia potential est [Internet]. Wikipedia; 2018 [cited 2018 July 17]. Available from: https://en.wikipedia.org/wiki/Scientia_potentia_est.
- Theendakara V, Peters-Libeu CA, Bredesen DE, Rao RV. Transcriptional Effects of ApoE4: Relevance to Alzheimer's disease. Mol Neurobiol. 2018; 55: 5243-5254. [CrossRef]
- Fossel M. Cells, Aging, and Human Disease. Oxford, New York: Oxford University Press; 2004.
- Personal communication - Michael Fossel.
- Weindruch R. The retardation of aging by caloric restriction: studies in rodents and primates. Toxicol Pathol. 1996; 24: 742-745. [CrossRef]
- Kennedy BK, Steffen KK, Kaeberlein M. Ruminations on dietary restriction and aging. Cell Mol Life Sci. 2007; 64: 1323-1328. [CrossRef]
- Ingram DK, Roth GS. Calorie restriction mimetics: can you have your cake and eat it, too? Ageing Res Rev. 2015; 20C: 46-62. [CrossRef]
- Ingram DK, Zhu M, Mamczarz J, Zou S, Lane MA, Roth GS, et al. Calorie restriction mimetics: an emerging research field. Aging Cell. 2006; 5: 97-108. [CrossRef]
- Pistollato F, Sumalla Cano S, Elio I, Masias Vergara M, Giampieri F, Battino M. Associations between sleep, cortisol regulation, and diet: Possible implications for the risk of Alzheimer disease. Adv Nutr. 2016; 7: 679-689. [CrossRef]
- Rosenberg A, Ngandu T, Rusanen M, Antikainen R, Bäckman L, Havulinna S, et al. Multidomain lifestyle intervention benefits a large elderly population at risk for cognitive decline and dementia regardless of baseline characteristics: The FINGER trial. Alzheimers Dement. 2018; 14: 263-270. [CrossRef]
- Yuede CM, Timson BF, Hettinger JC, Yuede KM, Edwards HM, Lawson JE, et al. Interactions between stress and physical activity on Alzheimer's disease pathology. Neurobiol Stress. 2018; 8: 158-171. [CrossRef]
- Hölscher C. First clinical data of the neuroprotective effects of nasal insulin application in patients with Alzheimer's disease. Alzheimers Dement. 2014; 10: S33-S37. [CrossRef]
- Claxton A, Baker LD, Wilkinson CW, Trittschuh EH, Chapman D, Watson GS, et al. Sex and ApoE genotype differences in treatment response to two doses of intranasal insulin in adults with mild cognitive impairment or Alzheimer's disease. J Alzheimers Dis. 2013; 35: 789-797. [CrossRef]
