

Epigenetic Symmetry of DLGAP2: Pre-Implantation Maternal Methylation Switches to a Random Monoallelic Profile in Somatic Tissues
Ana Monteagudo-Sánchez 1,†![]() , Marta Sánchez-Delgado 1,†
, Marta Sánchez-Delgado 1,†![]() , Sonia Guara Ciurana 1,†
, Sonia Guara Ciurana 1,†![]() , Jose Medrano 2,‡
, Jose Medrano 2,‡![]() , Maria Eugenia Poo-Llanillo 3
, Maria Eugenia Poo-Llanillo 3![]() , Miho Ishida 4
, Miho Ishida 4![]() , Gudrun E. Moore 4
, Gudrun E. Moore 4![]() , Isabel Iglesias-Platas 5
, Isabel Iglesias-Platas 5![]() , Carlos Simón 3,6,7
, Carlos Simón 3,6,7![]() , David Monk 1,*
, David Monk 1,*![]()
- Cancer Epigenetics and Biology Program (PEBC), Bellvitge Institute for Biomedical Research (IDIBELL), Barcelona, Spain
- Fundación IVI-Instituto Universitario IVI- INCLIVA, Valencia, Spain
- Igenomix SL, Valencia, Spain
- Great Ormond Street Institute of Child Health (ICH), University College London (UCL), London, UK
- Neonatal Service, Institut de Reçerca Sant Joan de Déu, Center for Neonatal & Maternofetal Medicine Barcelona (BCN-Natal), University of Barcelona, Spain
- Department of Obstetrics & Gynaecology, Valencia University, Valencia, Spain.
- Department of Obstetrics & Gynaecology, Stanford University, Palo Alto, California, USA.
‡ Current Affiliation: Instituto de Investigación Sanitaria La Fe, Valencia, Spain
† These authors contributed equally to this work.
* Correspondence: David Monk![]()
Received: May 29, 2018 | Accepted: July 11, 2018 | Published: July 30, 2018
OBM Genetics 2018, Volume 2, Issue 3 doi:10.21926/obm.genet.1803026
Academic Editors: Stéphane Viville and Marcel Mannens
Special Issue: Epigenetic Mechanisms in Health and Disease
Recommended citation: Sánchez AM, Delgado MS, Ciurana SG,Medrano J, Poo-Llanillo ME, Ishida M, Moore GE, Platas II,Simón C, Monk D. Epigenetic Symmetry of DLGAP2: Pre-Implantation Maternal Methylation Switches to a Random Monoallelic Profile in Somatic Tissues. OBM Genetics 2018;2(3):026; doi:10.21926/obm.genet.1803026.
© 2018 by the authors. This is an open access article distributed under the conditions of the Creative Commons by Attribution License, which permits unrestricted use, distribution, and reproduction in any medium or format, provided the original work is correctly cited.
Abstract
Background: Symmetrical DNA methylation profiles of autosomal genes are associated with equal expression by both alleles. Genes with an allelic imbalance or monoallelic expression are associated with discrete intervals of allele-specific methylation (ASM), as highlighted by genomic imprinting, X-chromosome inactivation and genotype-driven ASM. However, a more complex pattern has been described in which random monoallelic methylation provides cells with a unique mechanism for modulating allelic dosage. Methods: We combined direct interrogation of genome-wide methyl-seq datasets with locus-specific methylation with expression analysis to characterize the epigenetic profile of a CpG island associated with the DLGAP2 gene. The random nature of the ASM was confirmed using both bisulfite PCR in tissues and single cell-derived clonal analysis. Results: We identified an interval of oocyte-derived methylation manifested as a maternally methylated differentially methylated region (DMR) in human blastocysts and placenta. This switched to a random ASM profile by week 16 of gestation. Characterization using 5’ RACE-PCR revealed linkage of the ERICH1-AS1 transcript to DLGAP2, presenting an alternative transcription start site. Quantitative RT-PCR of DLGAP2 suggested a highly restricted expression profile limited to testis and brain, with allelic RT-PCR demonstrating robust biallelic expression. Conclusions: While many intervals subject to transient maternal methylation in the human pre-implantation embryo resolve to a fully unmethylated state in somatic tissues, we describe the first example of a CpG island converting to a random ASM profile. This profile has parallels with X-chromosome inactivation (XCI) in female mice, in which XCI is initially imprinted during pre-implantation development and maintained in the placenta, while derivatives of the inner cell mass are subject to random XCI. DLGAP2 has been associated with many neurological disorders, indicating a potential role of allele-specific expression and random ASM in the presentation of the disease phenotypes.
Graphical abstract
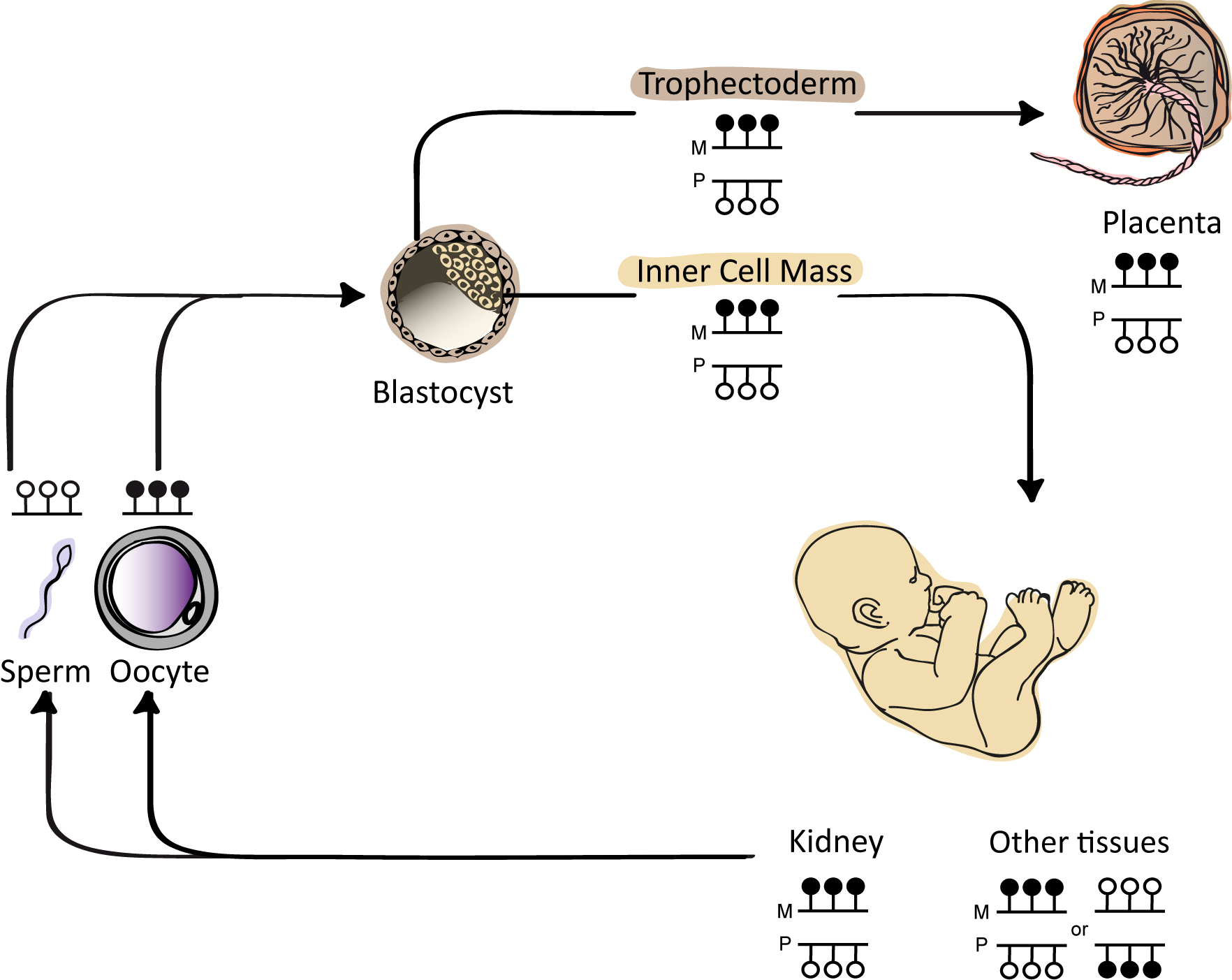
Keywords
Epigenetics; imprinting; allele-specific methylation; germline methylation; DLGAP2
1. Introduction
Allele-specific methylation (ASM) is a hallmark of several distinct epigenetic phenomena. One of the best characterized is genomic imprinting, where discrete intervals of germline-derived methylation survive embryonic reprograming, resulting in parent-of-origin monoallelic expression [1]. The CpG dinucleotides within imprinted differentially methylated regions (DMRs) are methylated on one parental allele, whereas the other is unmethylated, resulting in the characteristic monoallelic expression. A second mechanism associated with monoallelic methylation is X-chromosome inactivation in female mammals, in which one of the two X-chromosomes is randomly inactivated in somatic cells to compensate gene expression between XY males and XX females [2]. These results in female tissues being a mosaic of cells with reciprocal X-chromosome inactivation (XCI) patterns that display clonally inherited allelic methylation. However, a surprisingly large number of autosomal loci are also subject to stochastic ASM, with some, but not all being associated with random monoallelic expression [3]. This third class of genes is expressed in a random and clonal fashion from either the paternal or maternal allele, in a manner similar to random XCI but unlike imprinting. This category of genes includes members of large gene families, such as olfactory receptors [4], protocadherins [5] and immunoglobins [6], the expression of which is usually highly specific.
In addition to the classic epigenetic mechanisms (i.e. the methylation is classically independent of the underlying genetic sequence) mentioned, genotype-dependent ASM has been described for which genetic variants act in cis to dictate methylation of neighboring CpG positions. These discrete regions, referred to as methylation quantitative trait loci (meQTLs), are often tissue-specific [7,8] and have been hypothesized to contribute to inter-individual phenotype variation[8,9,10]. There is strong evidence suggesting that variations in methylation patterns at regulatory elements influence neurodevelopmental processes and the complex pathways involved in brain disorders with several studies revealing a link between cis-regulatory meQTLs and susceptibility loci identified in single nucleotide polymorphism (SNP) association studies [11,12].
In a recent screen for germline-derived methylation differences that survive embryonic reprogramming and persist in the blastocyst, we identified a CpG island associated with the DLGAP2 gene, encoding the large (Drosophila) homolog-associated protein 2, on chromosome 8 as a potential maternally methylated imprinted DMR [13]. This gene was also identified during a computational screen for loci with genomic features enriched at imprinted loci and was reported to be paternally expressed in fetal testis tissue, in a manner that was consistent with genomic imprinting [14]. Interestingly, cytogenetic studies have revealed that copy-number variations (CNVs) of the 8p23.3 interval are associated with many neurological and psychiatric disorders, including autism spectrum disorder (ASD) [15], schizophrenia [16] and Alzheimer’s disease [17], with specific DLGAP2 genotypes/haplotypes over-represented in patients compared to controls [18]. This suggests a potential role for the DLGAP2 gene in these disorders.
In this study, we have characterized the epigenetic profile of the DLGAP2 intergenic DMR using allele-specific analyses to investigate DNA methylation, histone modifications and gene expression in fetal and adult tissues as well as single cell-derived lymphoblastoid isogenic clones. We identified differential methylation of the DLGAP2 DMR between gametes and showed that the parent-of-origin methylation was maintained throughout pre-implantation development and in the placenta and kidney; all other tissues displayed random monoallelic methylation. Importantly, the allelic methylation was shown to be concomitant with an opposing permissive chromatin state, with the unmethylated alleles being enriched for methylation of lysine 4 of histone H3 (H3K4me). The shared features with XCI in mice [19] indicate that DLGAP2 is an example of an autosomal locus controlled by an XCI-like mechanism.
2. Material and Methods
2.1. Samples
Peripheral blood was obtained from healthy volunteers or from the umbilical cord of newborns for which matched placental biopsies were obtained. These samples were collected at the Hospital St. Joan De Déu (Barcelona, Spain). For the 48 placenta-derived DNA samples, microsatellite repeat analysis confirmed the absence of maternal DNA contamination. The adult brain samples were obtained from BrainNet Europe/Barcelona Brain Bank and adult somatic tissue samples were obtained from the Catalonia Tissue Bank Network. The dissection of individual brain regions was performed by an experienced pathologist on cadavers within 14 hours of death. A total of 48 fetal tissue sets (8–18 weeks of gestation), the majority with corresponding maternal blood samples, were obtained from the termination of pregnancies at Queen Charlotte’s and Chelsea Hospital, (London, UK). Twelve human metaphase II oocytes and a single surplus human blastocyst were obtained from the Instituto Valenciano de Infertilidad (FIVI) (Valencia, Spain). Vitrified oocytes were thawed using a kit for de-vitrification (Kitazato Valencia, Spain) following manufacturer’s instructions.
2.2. Methods
Genotyping and Imprinting Analysis. Genotypes of potential SNPs identified in the UCSC hg19 browser (https://genome.ucsc.edu) were obtained by PCR and direct sequencing. PCR amplicons were precipitated with ethanol to remove excess primers. Approximately 30 ng of clean PCR product was used as template in a sequence reaction using BigDye terminator master mix (Applied Biosystems, now Thermo Fisher Scientific, Massachusetts, USA) and separated using capillary electrophoresis (Applied Biosystems Genetic Analysis System, ABI 3730 DNA Analyzer). Sequence traces were interrogated using Sequencher v4.6 (Gene Codes Corporation, MI, USA) to distinguish heterozygous and homozygous samples. Heterozygous tissue samples were analyzed for either allelic expression by RT-PCR or bisulfite PCR, incorporating the polymorphism within the final PCR amplicon to facilitate identification of the parental alleles (for primer sequences see Supp. Table S1).
Methylation-Sensitive Genotyping. Approximately 500 ng of heterozygous genomic DNA was digested with 10 units of HpaII restriction endonuclease (NEB, Massachusetts, USA) for 6 hours at 37°C. The digested DNA was isolated by ethanol precipitation and resuspended in a final volume of 20 μl TE or water. A sample (2 μl) of the digested DNA was used in each PCR amplification (initial denaturation 96°C for 5 minutes; 40 cycles of denaturation at 96°C 30 seconds, 56°C annealing for 30 seconds and 72°C extension for 1 minute; final extension 5 minutes at 72°C) using Taq polymerase (Bioline, London, UK). The resulting amplicons were sequenced and the traces compared to those obtained for the corresponding undigested DNA template.
Rapid Amplification of cDNA ends (RACE) PCR. 5’ RACE-PCR was used to obtain the full-length sequence of the DLGAP2 mRNA transcript using the 5’/3’ RACE kit (Roche, Basel, Switzerland) according to the manufacturer’s instructions.
Quantitative Real-Time-PCR. Expression of DLGAP2 and ERICH1-AS1 transcripts was analyzed using a fluorochrome (SYBR® Green)-based quantitative real-time RT-PCR assay and normalized against RPL19. cDNA was synthesized as previously described [20]. All assays were run in triplicate in 384-well plates using the 7900HT Fast Real-Time-PCR System (Applied Biosystems, California, USA). Only samples with two or more valid readings per triplicate were included in our analysis. Dissociation curves were generated at the end of each reaction to rule out the presence of primer-dimers or unexpected DNA species in the reaction. Non-template controls, an inter-plate control and standard curves generated using the same serial dilutions of cDNA obtained from pooled normal tissue were included in each assay. Results were analyzed with the SDS 2.3 software (Applied Biosystems).
Allele-Specific Bisulfite PCR. For standard bisulfite conversion, we used the EZ DNA Methylation-Gold kit (ZYMO, California, USA) according to the manufacturer’s instructions. Approximately 2 µl of bisulfite-converted DNA was used in each amplification reaction (initial denaturation 96°C for 10 minutes; 45 cycles of denaturation at 96°C 30 seconds, 53°C annealing for 30 seconds and 72°C extension for 1 minute; final extension 5 minutes at 72°C) catalyzed by Immolase Taq polymerase (Bioline). The resulting PCR product was sub-cloned into the pGEM-T easy vector (Promega, Southampton, UK) for sequencing (for primer sequences see Supp. Table S1). The EZ DNA Methylation-Direct kit (ZYMO) (in which cell lysates are subject to direct bisulfite conversion) was used for the methylation profiling of individual hair follicles, the blastocyst and pooled oocytes. Nested bisulfite PCR was required to obtain amplicons for subcloning due to the small amounts of available samples.
Chromatin Immunoprecipitation Assay. The allelic enrichment of histone modifications in peripheral leukocytes, placenta and brain samples was confirmed by chromatin immunoprecipitation (ChIP) as previously described [21], with minor modifications. For blood samples, mononuclear cells were first isolated by Lymphoprep (Stem Cell Tech, Vancouver, Canada) density gradient centrifugation, while 100 mg of tissue was used for placenta and brain. Washed samples were disrupted to release nuclei using 0.5 mM zirconium beads (Sigma–Aldrich, Missouri, USA) in a Precellys 24 tissue homogenizer (Bertin_Corp, Maryland, USA) and subsequently isolated by centrifugation (at 13,000g with centrifuge set on slowest deceleration rate) for use in micrococcal nuclease (MNase) digestion. Approximately 4 µg of chromatin was used for an immunoprecipitation reaction with protein A agarose/salmon sperm DNA (Thermofisher) and antibodies specific for H3K4me3 (C15410003-50 Diagenode, Seraing, Belgium), H3K4me2 (07-030 Millipore,Massachusetts, USA) and H3K9me3 (AB8898 Abcam, Cambridge, UK). A mock immunoprecipitation with an unrelated IgG antiserum (12-371, Millipore) was performed in parallel with each ChIP, and a 50% fraction of the input chromatin was extracted in parallel. The precipitated DNA was subjected to column-based clean-up (Macherey-Nagel, Barcelona, Spain) and 2 µl was used for allelic PCR amplification.
For quantitative analysis, the input and antibody-bound fractions were subjected to real-time PCR amplification (initial denaturation 95°C for 10 minutes followed by 40 cycles of 95°C 15 seconds and 60°C extension for 1 minute) with a SYBR Green mixture (SYBR® Green) using a 7900HT Fast Real Time PCR (Applied Biosystems) instrument. Background precipitation levels were determined using the mock precipitations. Bound/input ratios were calculated and normalized against the precipitation level at the GAPDH promoter for active marks and SAT2 repeats for H3K9me3. All PCRs were performed in triplicate and the primers used are shown in Supplementary Table S1.
Single Cell Lymphoblastoid Colony Expansion. Isolated clonal cell lines were generated by dilution plating of a polyclonal lymphoblastoid cell line at a ratio of 0.4 cells per well. Transformation with Epstein–Barr virus (EBV) was performed for a control individual who was heterozygous for SNPs within the DLGAP2 ASM. The B-cells within the sample were immortalized with the supernatant of the EVB producer cell line B95.8. Surviving colonies were propagated to a density sufficient for DNA isolation. Ethical approval for this study was granted by the Institutional Review Boards at the Bellvitge Institute for Biomedical Research (PR006/08 & PR096/10) and the Ethics Committee of the Hospital St. Joan De Déu (35/07). Local ethical approval for obtaining fetal tissues was granted by the Research Ethics Committee of the Hammersmith, Queen Charlotte’s and Chelsea and Acton Hospitals (2001/6028) and the use of surplus human embryos and gametes was evaluated and approved by the scientific and ethics committees of the Instituto Valenciano de Infertilidad (IVI) (1310-FIVI-131-CS), the Bellvitge Institute for Biomedical Research (PR292/14), the National Committee for Human Reproduction (CNRHA) and the Regional Health Counsel of Valencia. Written informed consent was obtained from all participants.
3. Results
3.1. Defining the Extent of ASM of DLGAP2
Using methyl-seq datasets from gametes, pre-implantation embryos and various tissues representative of the three germ layers, we confirmed that a CpG island (hg19 chr8:1321233-1321638, USCS identifier CpG: 46; see reference [13] for information related to the bioinformatic screening and datasets used), located between ERICH1-AS1 and DLGAP2 was fully methylated in oocytes and unmethylated in sperm (Figure 1A-B). The unmethylated interval in sperm extended for approximately 1.5 kb and overlapped with a partially methylated region in blastocysts. In somatic tissues and placenta, the region of partial methylation defining the potential imprinted DMR was shorter, at approximately 550 bp (hg19 chr8:1321145-1321758). Importantly however, interrogation of methyl-seq datasets only provides information regarding absolute methylation of each CpG within the region and does not discriminate between the various forms of ASM. Using a strategy combining nested bisulfite PCR with subcloning, we confirmed that human oocytes were fully methylated, while sperm was devoid of methylation. Similar analysis performed on a single human day 5 blastocyst revealed strand-specific methylation, which was present on only one allele, as this sample was heterozygous for the SNP rs36018196 (Figure 1C).
3.2 Allele-Specific Methylation Analysis in Placenta
To confirm restriction of the observed methylation to the maternal allele in the final stage of development, we established a methylation-sensitive genotyping assay. This method involved polymorphic allele-calling on genomic DNA before and after digestion with the methylation-sensitive HpaII endonuclease (Figure 2A; Supplementary Figure 1). Allelic methylation is confirmed when a heterozygous genomic DNA sample is reduced to homozygosity following digestion with the remaining allele representing the methylated chromosome. We initially genotyped eight SNPs within the regions; however, only two were consistently informative (rs6996211 and rs36018196). We have previously shown that the regions of maintained oocyte-derived methylation are frequently maternally methylated in the placenta [13]; therefore, we focused our initial studies on this extra-embryonic tissue. In total, 18 samples were informative for the two SNPs allowing determination of the parental origin of methylation. In all cases, we confirmed monoallelic methylation, with four being cases shown to be methylated on the maternal allele when parental genotypes were available and informative (Figure 2B). To confirm that the allelic methylation was not restricted to the CpG dinucleotides contained in the 5 HpaII sites within the amplified interval, we performed bisulfite PCR. The results revealed that the ASM encompassed at least 23 CpGs within the 157-bp amplified region (Figure 2C).
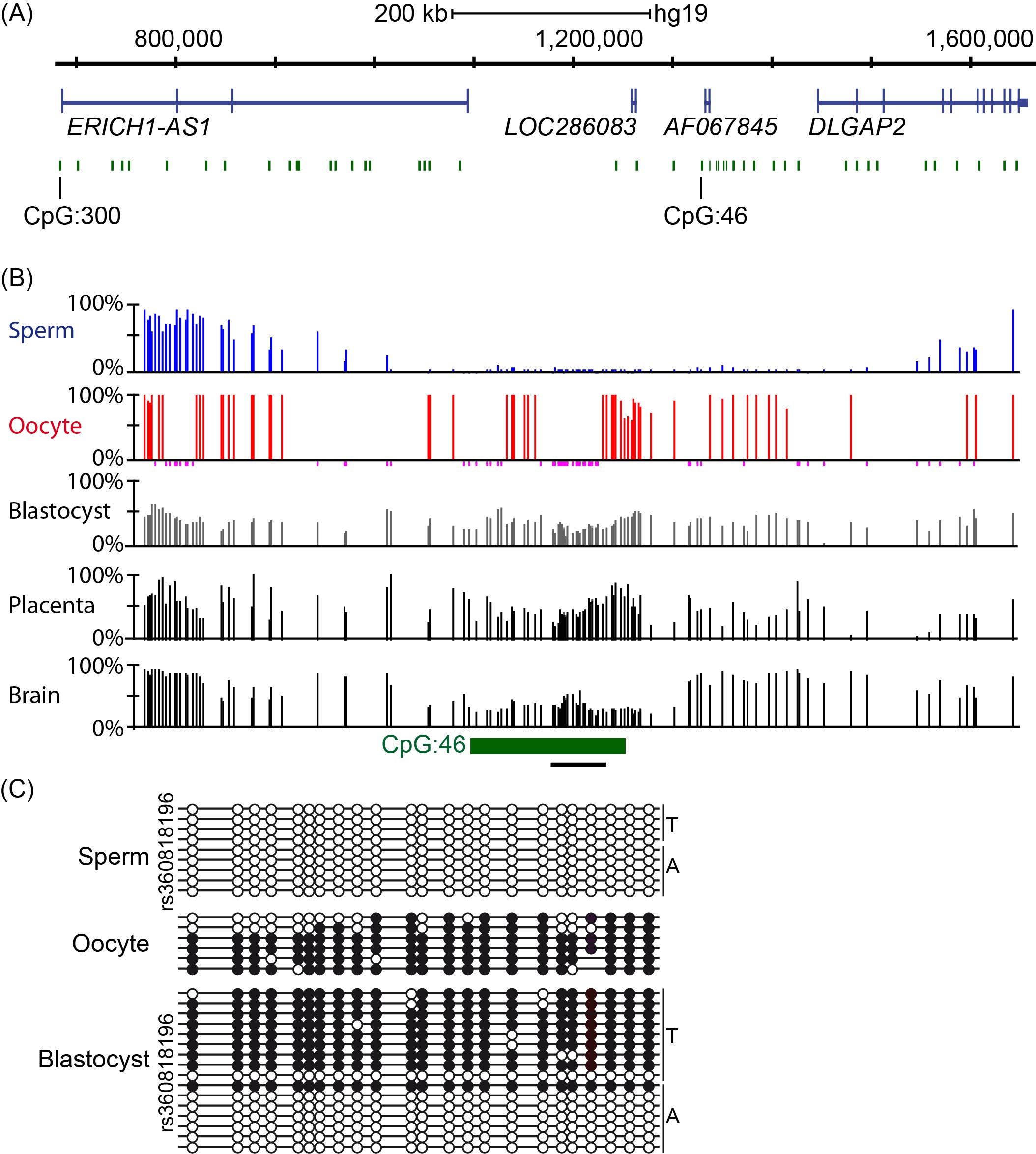
Figure 1 Methylation analysis of CpG island 46 upstream of DLGAP2. (A) Genomic map of the Chr8p23.3 interval containing ERICH1-AS1 and DLGAP2. CpG islands within this interval of approximately 983 kb are shown in green and the exons in each transcript are shown in blue. (B) Methyl-seq datasets showing that CpG 46 is unmethylated in sperm, fully methylated in oocytes and partially methylated in blastocysts, placenta and somatic tissues (only brain shown). The vertical lines in the methyl-seq tracks represent the mean methylation values for individual CpG dinucleotides. The black line under CpG:46 indicates the localization of the bisulfite PCR product analyzed in panel C. (C) Bisulfite PCR and subcloning was used for confirmation. Each circle represents a single CpG on a DNA strand. (•) Methylated cytosine, (o) unmethylated cytosine. Each row corresponds to an individual cloned sequence with the parent-of-origin indicated by the genotype of the heterozygous SNP incorporated into the amplicon.
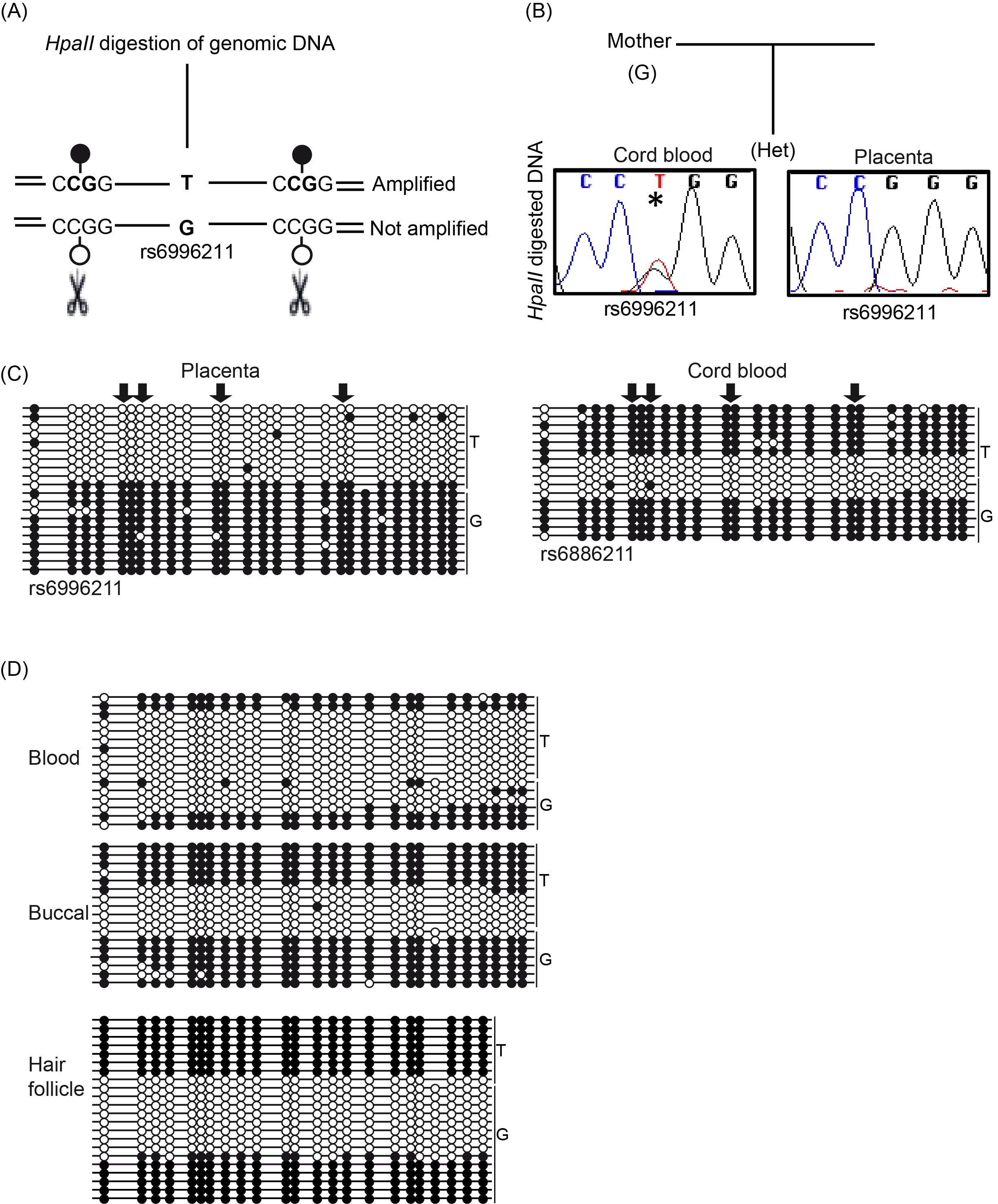
Figure 2 Allele-specific methylation analysis of DLGAP2. (A) Schematic overview of the methylation-sensitive HpaII genotyping assay and (B) the resulting methylation profile in paired cord blood and placenta samples. (C) Bisulfite PCR and subcloning was used for confirmation of the samples shown in panel B. The arrows indicate the position of the HpaII sites. (D) Additional bisulfite PCR and subcloning was performed on three DNA samples derived from a single individual revealing the stability of the ASM. Each circle represents a single CpG on a DNA strand. (•) Methylated cytosine, (o) unmethylated cytosine. Each row corresponds to an individual cloned sequence with the parent-of-origin indicated by the genotype of the heterozygous SNP incorporated into the amplicon.
3.3 DLGAP2 Contains a Random ASM Region in Blood
We subsequently performed bisulfite PCR and sub-cloning assays on cord blood derived DNA from the heterozygous placenta samples. In all cases, the methylation was strand-specific, although allelic discrimination using the informative SNPs revealed that both the maternally- and paternally-derived alleles were a mix of fully methylated and unmethylated strands consistent with random monoallelic ASM (Figure 2C). To investigate the stability of this bimodal profile during development, we investigated the allelic methylation in adult leukocytes. Similarly, we observed random monoallelic ASM in two individuals, with identical profiles detected in accompanying saliva and hair follicle-derived DNA (Figure 2D).
To further investigate this pattern of methylation, we performed methylation-sensitive genotyping on single cell-derived clones. Lymphoblastoid cells derived from one of the informative adult blood donors were generated by Epstein–Barr virus transformation and homogenous lines derived from single cells were propagated. In the unsorted bulk population, the rs6996211 SNP remained heterozygous following HpaII digestion, indicating that methylation was randomly present on both alleles. However, of the 21 single cell derived clones that were successfully generated, 12 were monoallelically methylated on the paternally-derived G allele and seven on the opposite maternal T allele (Figure 3).
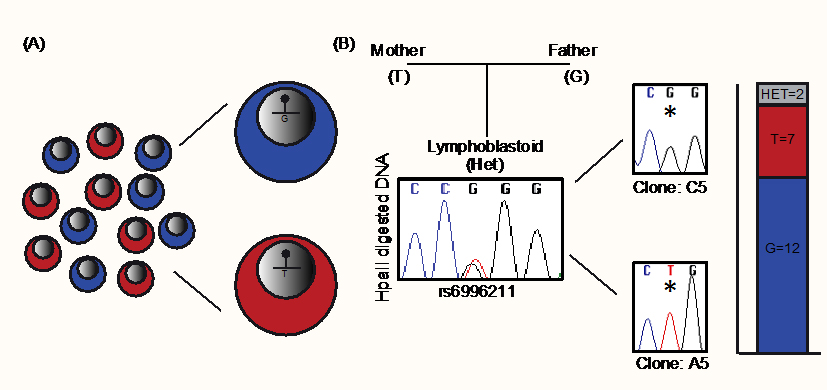
Figure 3 Characterization of DLGAP2 ASM through analysis of single cell-derived clones. (A) Schematic illustration of the experimental design. Single cells were isolated from a lymphoblastoid cell line that was heterozygous for SNP rs6996211 and expanded to derive reciprocal isogenic clones. (B) Methylation-sensitive HpaII genotyping reveals biallelic methylation of the interval in an unsorted “bulk” sample, whereas isogenic clones were monoallelic in 90% of cases. The bar graph shows the number of individual clones with random monoallelic methylation.
3.4 Random ASM in DLGAP2 is Stable in Somatic Tissues
We next aimed to further characterize the methylation profile in various fetal and adult tissues derived from the three germ layers (Figure 4A). Following genotyping of 48 tissue sets and corresponding maternal blood, we identified a single fetus that was informative for SNP rs36018196, although multiple unrelated adult tissues were heterozygous for rs6996211 and rs36018196.
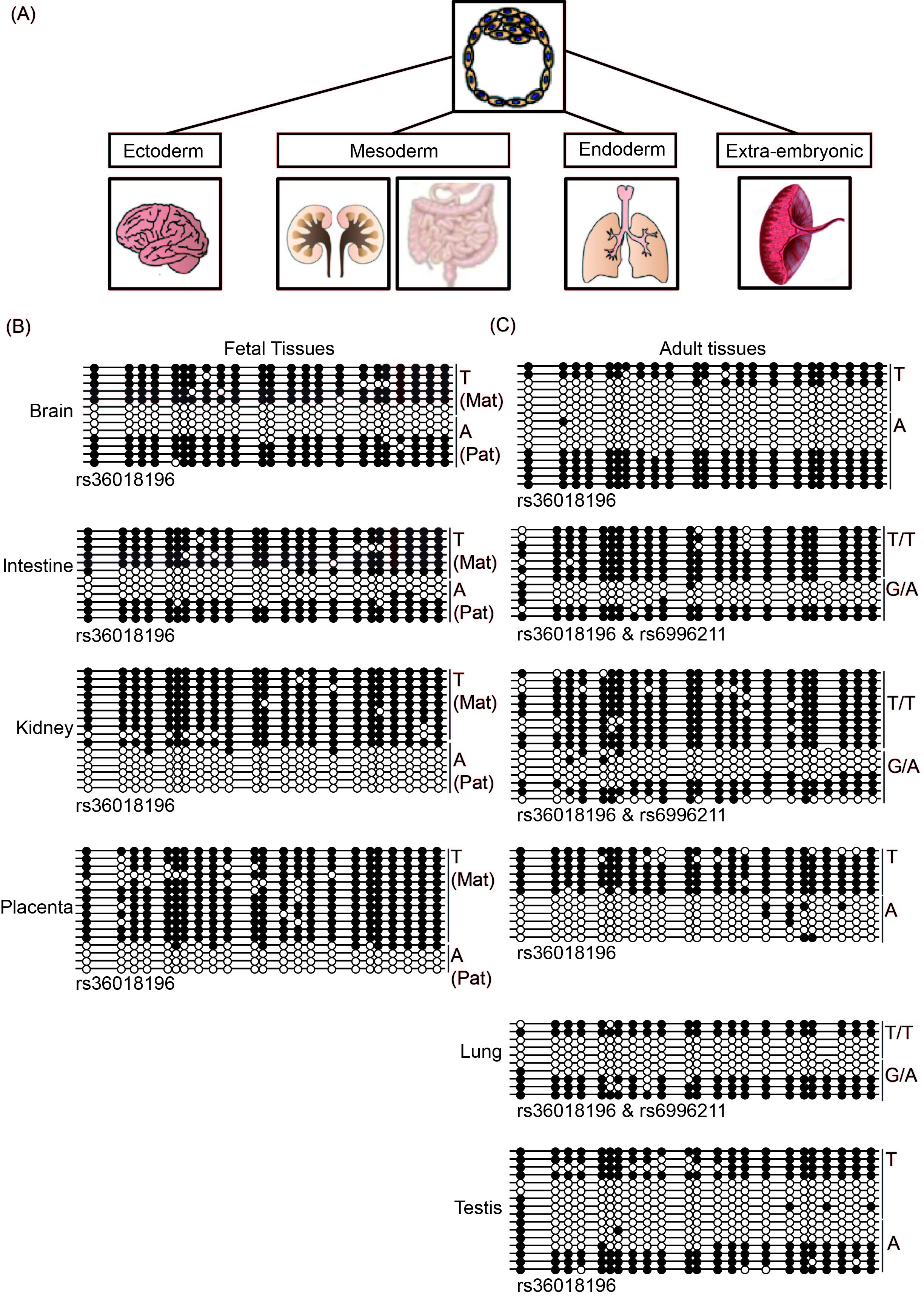
Figure 4 Defining the DLGAP2 ASM in multiple tissues at different developmental time-points.(A) Schematic diagram showing the tissues used and their germ layer origins. (B) Characterization of the methylation profiles in four tissues from a fetal sample set informative for SNP rs36018196 by bisulfite PCR and subcloning. (C) An identical approach was used to study the ASM interval in unrelated adult tissues and term placenta. Note that in some cases the samples were heterozygous for both rs36018196 and rs6996211 SNPs.
We performed bisulfite PCR and subcloning on four tissues from the informative fetus (approximately 16 weeks of gestation). In accordance with previous findings, we observed that the methylation in the placenta was restricted to the maternal allele. Surprisingly, a similar profile was also observed in kidney, while both brain and intestine tissues were subject to random ASM (Figure 4B). In adult tissues, random ASM was clearly observed in lung and testis as well as the frontal cortex and hippocampus of the brain (Figure 4C; Suppl Figure 1). The only informative adult intestine sample was heterozygous for both SNPs and fully methylated on the T/T allele, while a mix of methylated and unmethylated strands was detected on the G/A allele. Finally, although not as clearly defined, one allele in adult kidney was preferentially methylated; however,the parental origin could not be ascertained because of the lack of accompanying parental samples.
3.5 The DLGAP2 ASM Region is Decorated with Active Histone Markers
Histone modifications are another potential distinguishing epigenetic feature of regions subject to ASM. Opposing active and repressive chromatin states are commonly observed for the inactive X-chromosome and at imprinted DMRs, but very little is known about other forms of ASM or meQTLs. To investigate the underlying chromatin state in the DLGAP2 DMR, we analyzed H3K4 di- and trimethylation as markers of permissive chromatin and H3K9me3 as markers of repressive chromatin. In the placenta, we observed strong immunoprecipitation of H3K4me3 and residual H3K4me2, both of which were enriched on the paternal allele, while no allelic bias was observed for H3K9me3 (Figure 5A-B). In accordance with the random nature of the ASM in blood and brain, we failed to observe allelic enrichment of any histone modification in “bulk” samples of adult leukocytes or frontal cortex tissue (Figure 5 C-D).
3.6 DLGAP2 is a Biallelically Expressed Gene
We interrogated mRNA and expressed sequence tags (EST) to identify transcripts within the vicinity of DLGAP2 that may originate near the DMR. This revealed that transcript AK067845 originates in a region close to the ASM interval. Unfortunately, we were unable to detect expression of these exons. To determine if the DMR represents a promoter for a yet-to-be reported isoform, we performed 5’ RACE experiments. The results revealed that the furthest upstream transcriptional start site (TSS) for DLGAP2 originated from within the unmethylated CpG island and had identical sequence similarity to EST BC022082, which was annotated as ERICH1-AS1 (Suppl Figure 2). Comparisons with other mammalian species, including rat and mouse, revealed that Erich1-as1 was a bona fide alternative TSS for Dlgap2. In fact, during this study, the Ensembl annotation of ERICH1-AS1 (Gene ID: 619343) was replaced by DLGAP2 (Gene ID: 9228), which endorsed our observations. To provide additional evidence of the linkage between the ERICH1-AS1 exons and the DLGAP2 gene, we performed RT-PCR analysis of the two transcripts in brain and testis tissues. A single PCR amplicon was obtained that was shown the contain the expected exon splicing junctions in addition to a novel exon in sequencing analysis (Figure 6A). Furthermore, qRT-PCR analysis of the regions between exons 1–2 of ERICH1-AS1 and exons 6–8 of DLGAP2 revealed highly comparable expression profiles, being most abundant in testis and in the putamen and frontal and occipital cortices of the brain; these results were consistent with the profiles in GTEx datatsets (Figure 6B; data not shown). No expression was detected in the placenta. Finally, to determine the allelic expression of DLGAP2, we performed RT-PCR analysis of the regions spanning the exonic SNPs rs4565482 and rs2235112 located in exon 6. The results showed that the gene is robustly expressed biallelically in the frontal cortex, thalamus, bulb, cerebellum and hippocampus of 12 different brain samples (Figure 6C). In accordance with ERICH1-AS1 being upstream of DLGAP2, RT-PCR analysis targeting these exons also revealed equal expression from both parental alleles for SNP rs141356800 in adult brain and testis samples.

Figure 5 Profiling of histone modifications associated with the DLGAP2 ASM interval. (A) Quantitative chromatin analysis following native ChIP using anti-H3K4 and H3K9 antibodies. The precipitation levels were normalized to those obtained for the GAPDH promoter (for H3K4) and the SAT2 repeat (for H3K9). Allelic distribution of each histone modification was determined by direct sequencing of the PCR amplicons encompassing heterozygous SNPs for (B) placenta, (C) leukocytes and (D) frontal cortex of the brain.
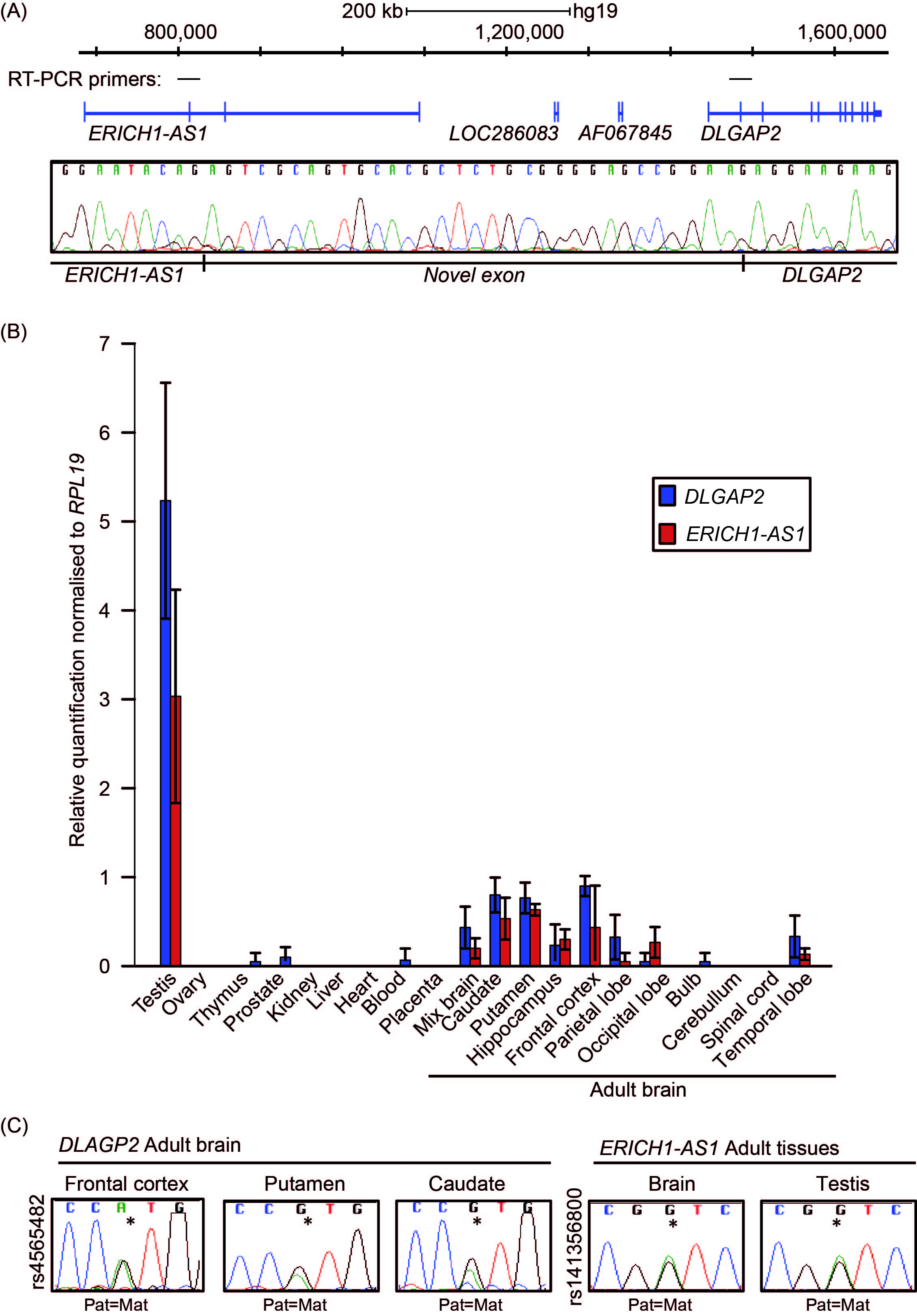
Figure 6 Allele-specific expression of the DLGAP2 gene. (A) Schematic overview of the ERICH1-AS1 and DLGAP2 genes and the positions of the primers used for linking the two transcripts. The electropherogram reveals the sequence of the novel exon located between the two transcripts. (B) Quantitative RT-PCR showing the distribution of expression across different tissues. The mean values are shown with 95% CI. (C) Sequence traces of RT-PCR products for DLGAP2 and ERICH1-AS1 in adult tissue samples heterozygous for SNPs rs4565482 and rs141356800, respectively.
4. Discussion
The monoallelic expression of non-imprinted autosomal genes is not a sporadic phenomenon, being observed for approximately 10% of genes in various cell types [22,23]. However, the mechanism dictating stochastic allelic choice remains elusive and is likely to involve several epigenetic mechanisms. In the case of DLGAP2, we observed a methylation profile consistent with classical imprinting in the pre-implantation embryo, which was reinforced by H3K4 methylation of the unmethylated allele in a pattern that was similar to the canonical signature associated with imprinted DMRs [24,25]. However, the process by which the switchable random ASM pattern is established post-implantation is unclear.
Similar patterns of random monoallelic ASM have been observed in mouse embryonic stem (ES) cells. Recently, Martos and colleagues described a region near the Park7 gene in mice, for which half of the clones were methylated on the maternal and paternal alleles in ES lines derived from reciprocal F1 hybrids of C57BL/6 and Castaneous crosses [26]. This switchable feature of ASM resembles that of random inactivation of one of the two X chromosomes in females, leading the authors to propose that some autosomal loci are subject to autosomal chromosome inactivation (ACI), similar to XCI, and that silencing would be centred around a focal region of ASM [19,26]. In accordance with this hypothesis, the DLGAP2 ASM shows similarities with XCI in mice, in which random inactivation of one X-chromosome in females ensures dosage compensation between XX and XY. During this process, one of the two X chromosomes is silenced early in development and once established, the inactive state is stably maintained through mitosis by a combination of chromatin-remodeling proteins, histone variants and post-translational modifications, DNA methylation and asynchronous replication [27].
The results of this study highlight the importance of exhaustive studies of allelic methylation for correct annotation of the mechanism associated with the ASM. Strand-specific bimodal methylation patterns detected by bisulfite PCR and subcloning have long been used to validate imprinting; however, as shown in this example of DLGAP2, correctly assigning strand-specific methylation based on discriminating alleles using SNPs revealed the existence of both imprinted and random ASE in a tissue-specific fashion. Despite our best attempts to show concomitant monoallelic expression associated with DLGAP2 ASM, we could only discount imprinted expression. Unfortunately, DLGAP2 is not expressed in either the placenta or kidney; therefore, investigation of allelic expression with reproducible parent-of-origin methylation in these tissues was not possible. Biallelic DLGAP2 expression was observed in brain, which is consistent with the inheritance of cytogenetically defined deletions and duplications of 8q23.3 from either parent in patients with ASD and intellectual disabilities [28,29]; Decipher database]. Single cell RNA-seq or RNA-FISH analyses are required to confirm random monoallelic expression in brain since single cell clonal expansion is not currently feasible; however, it must be noted that the results from these techniques may not reflect mitotically stable random allelic expression because gene expression within single cells is associated with allelic bursts, which increases the risk of misinterpretation of monoallelic expression at a single time-point [30].
Random ASM results in random allelic expression in brain is likely to exert fine-tuning of gene dosage. Since this ASM would lead to somatic mosaicism, it is possible that this form of transcriptional regulation influences tissue composition, which could have advantages for organs such as the brain, causing a substantial impact on function. As mentioned previously, the protocadherin gene family is subject to random ASE and is implicated in self-recognition processes [31], which prevent the formation of self-contact by neurons and is essential for normal development of brain networks. Interestingly, DLGAP2, along with other DLGAP proteins, is involved in regulating transmission of neuronal signals across synaptic junctions and has been linked to a variety of neurological disorders, including ASD, schizophrenia, ASD, obsessive compulsive disorder (OCD) and Alzheimer’s disease [32]. Therefore, any mosaicism implying variability in gene expression due to random ASM could influence developmental plasticity, resulting in the incomplete penetrance of undesirable alleles and variable severity of the phenotype.
5. Conclusions
In this study, we characterized a discrete genomic interval associated with tissue-specific ASM. In a pre-implantation embryo and placenta, we observed robust methylation restricted to the maternal allele, while a random ASM profile was observed later in the development of tissues derived from all three germ layers. It has previously been proposed that imprinted loci have arisen from genes with random monoallelic expression that resulted in a selective advantage, with the ASM becoming fixed through evolution as a germline imprint [33]. It is, therefore, exciting to speculate that DLGAP2 is evolving to becoming an imprinted gene. Furthermore, since DLGAP2 has been implicated in several neurological disorders, any influence of the ASM on expression could have important implications for disease progression and severity due to the resulting inherent mosaicism.
Acknowledgments
We would like to thank all participants/donors in this study and the clinical staff involved in their care, especially the perinatal nurses at Hospital Sant Joan de Déu who contributed to the collection of control placental/cord blood samples. We thank the BrainNet Europe and the Catalonia Network of Tumor Banks (XBTC) for supplying normal tissues. Work supported by the Xarxa de Bancs de Tumors de Catalunya is sponsored by Pla Director d’Oncología de Catalunya.
Additional Materials
The following additional materials are uploaded at the page of this paper.
1. Supp. Figure 1. Methylation-sensitive genotyping of CpG: 46 of DLGAP2.
2. Supp. Figure 2. Methylation analysis of the ERICH1-AS1 promoter.
3. Supp. Table 1. Primer sequences used for PCR in this study.
Author Contributions
AMS, MSD and DM conceived and designed the experiments; AMS, MSD and SGC performed all the molecular and epigenetic characterizations; JM, MEPL and CS collected and processed the human gametes and blastocysts; IIP was responsible for the collecting and processing placenta and cord blood samples; MI and GEM genotyped the fetal and maternal DNA samples; DM drafted the manuscript. All authors contributed to the final revised manuscript version.
Funding
The laboratory of DM is supported by the Spanish Ministerio de Educación y Competitividad (BFU2014-53093 & BFU2017-85571-R) and co-funded by the European Union Regional Development Fund (FEDER). AMS is a recipient of a FPI PhD studentship from MINECO.
Competing Interests
The authors have declared that no competing interests exist.
References
- Monk D. Germline-derived DNA methylation and early embryo epigenetic reprogramming: The selected survival ofimprints. Int J Biochem Cell Biol. 2015; 67: 128-138. [CrossRef]
- Sharp AJ, Stathaki E, Migliavacca E, Brahmachary M, Montgomery SB, Dupre Y, et al. DNA methylation profiles of human active and inactive X chromosomes. Genome Res. 2011; 21: 1592-1600. [CrossRef]
- Eckersley-Maslin MA, Thybert D, Bergmann JH, Marioni JC, Flicek P, Spector DL. Random monoallelic gene expression increases upon embryonic stem cell differentiation. Dev Cell. 2014; 28: 351-365. [CrossRef]
- Magklara A, Yen A, Colquitt BM, Clowney EJ, Allen W, Markenscoff-Papadimitriou E, et al. An epigenetic signature for monoallelic olfactory receptor expression. Cell. 2011; 145: 555-570. [CrossRef]
- Kaneko R, Kato H, Kawamura Y, Esumi S, Hirayama T, Hirabayashi T, et al. Allelic gene regulation of Pcdh-α and Pcdh-γ clusters involving both monoallelic and biallelic expression in single Purkinje cells. J Biol Chem. 2006; 281: 30551-30560. [CrossRef]
- Bergman Y, Cedar H. A stepwise epigenetic process controls immunoglobulin allelic exclusion. Nat Rev Immunol. 2004; 4: 753. [CrossRef]
- Smith ZD, Chan MM, Humm KC, Karnik R, Mekhoubad S, Regev A, et al. DNA methylation dynamics of the human preimplantation embryo. Nature. 2014; 511: 611. [CrossRef]
- Marzi SJ, Meaburn EL, Dempster EL, Lunnon K, Paya-Cano JL, Smith RG, et al. Tissue-specific patterns of allelically-skewed DNA methylation. Epigenetics. 2016; 11: 24-35. [CrossRef]
- Kerkel K, Spadola A, Yuan E, Kosek J, Jiang L, Hod E, et al. Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation. Nat Genet. 2008; 40: 904-908. [CrossRef]
- Heyn H, Moran S, Hernando-Herraez I, Sayols S, Gomez A, Sandoval J, et al. DNA methylation contributes to natural human variation. Genome Res. 2013; 23: 1363-1372. [CrossRef]
- Schulz H, Ruppert A-K, Herms S, Wolf C, Mirza-Schreiber N, Stegle O, et al. Genome-wide mapping of genetic determinants influencing DNA methylation and gene expression in human hippocampus. Nat Commun. 2017; 8: 1511. [CrossRef]
- Sanchez-Mut JV, Heyn H, Silva BA, Dixsaut L, Garcia-Esparcia P, Vidal E, et al. PM20D1 is a quantitative trait locus associated with Alzheimer’s disease. Nat Med. 2018; 24: 598. [CrossRef]
- Sanchez-Delgado M, Vidal E, Medrano J, Monteagudo-Sánchez A, Martin-Trujillo A, Tayama C, et al. Human oocyte-derived methylation differences persist in the placenta revealing widespread transient imprinting. PLoS Genet. 2016; 12: e1006427. [CrossRef]
- Luedi PP, Dietrich FS, Weidman JR, Bosko JM, Jirtle RL, Hartemink AJ. Computational and experimental identification of novel human imprinted genes. Genome Res. 2007; 17: 1723-1730. [CrossRef]
- Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, Regan R, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010; 466: 368-372. [CrossRef]
- Li J-M, Lu C-L, Cheng M-C, Luu S-U, Hsu S-H, Chen C-H. Genetic analysis of the DLGAP1 gene as a candidate gene for schizophrenia. Psychiatry Res. 2013; 205: 13-17. [CrossRef]
- Chaudhry M, Wang X, Bamne MN, Hasnain S, Demirci FY, Lopez OL, et al. Genetic variation in imprinted genes is associated with risk of late-onset Alzheimer's disease. J Alzheimers Dis. 2015; 44: 989-994. [CrossRef]
- Chien W-H, Gau S-F, Liao H-M, Chiu Y-N, Wu Y-Y, Huang Y-S, et al. Deep exon resequencing of DLGAP2 as a candidate gene of autism spectrum disorders. Mol Autism. 2013; 4: 26. [CrossRef]
- Gendrel A-V, Marion-Poll L, Katoh K, Heard E, editors. Random monoallelic expression of genes on autosomes: Parallels with X-chromosome inactivation. Semin Cell Dev Biol; 2016; 56:100-110. [CrossRef]
- Iglesias-Platas I, Camprubi C, Sparago A, Guillaumet-Adkins A, Martin-Trujillo A, Riccio A, et al. Imprinting at the PLAGL1 domain is contained within a 70-kb CTCF/cohesin-mediated non-allelic chromatin loop. Nucleic Acids Res. 2013; 41: 2171-2179. [CrossRef]
- Tayama C, Romanelli V, Martin-Trujillo A, Iglesias-Platas I, Okamura K, Sugahara N, et al. Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of human imprinting and suggests a germline methylation-independent mechanism of establishment. Genome Res. 2014; 24: 554-569. [CrossRef]
- Gimelbrant A, Hutchinson JN, Thompson BR, Chess A. Widespread monoallelic expression on human autosomes. Science. 2007; 318: 1136-1140. [CrossRef]
- Savova V, Patsenker J, Vigneau S, Gimelbrant AA. dbMAE: the database of autosomal monoallelic expression. Nucleic Acids Res. 2015; 44: D753-D756. [CrossRef]
- Maupetit-Méhouas S, Montibus B, Nury D, Tayama C, Wassef M, Kota SK, et al. Imprinting control regions (ICRs) are marked by mono-allelic bivalent chromatin when transcriptionally inactive. Nucleic Acids Res. 2015; 44: 621-635. [CrossRef]
- Andergassen D, Dotter CP, Wenzel D, Sigl V, Bammer PC, Muckenhuber M, et al. Mapping the mouse Allelome reveals tissue-specific regulation of allelic expression. Elife. 2017; 6: e25125. [CrossRef]
- Martos SN, Li T, Ramos RB, Lou D, Dai H, Xu J-C, et al. Two approaches reveal a new paradigm of ‘switchable or genetics-influenced allele-specific DNA methylation’with potential in human disease. Cell Discov. 2017; 3: 17038. [CrossRef]
- Vallot C, Ouimette JF, Rougeulle C. Establishment of X chromosome inactivation and epigenomic features of the inactive X depend on cellular contexts. Bioessays. 2016; 38: 869-880. [CrossRef]
- Poquet H, Faivre L, Chehadeh SE, Morton J, Mcmullan D, Hamilton S. Further Evidence for Dlgap2 as strong Autism Spectrum disorders/intellectual disability candidate gene. Autism Open Access. 2016; 06.
- Guilmatre A, Dubourg C, Mosca A-L, Legallic S, Goldenberg A, Drouin-Garraud V, et al. Recurrent rearrangements in synaptic and neurodevelopmental genes and shared biologic pathways in schizophrenia, autism, and mental retardation. Arch Gen Psychiatry. 2009; 66: 947-956. [CrossRef]
- Raj A, van Oudenaarden A. Nature, nurture, or chance: stochastic gene expression and its consequences. Cell. 2008; 135: 216-226. [CrossRef]
- Kostadinov D, Sanes JR. Protocadherin-dependent dendritic self-avoidance regulates neural connectivity and circuit function. Elife. 2015; 4: e08964. [CrossRef]
- Rasmussen AH, Rasmussen HB, Silahtaroglu A. The DLGAP family: neuronal expression, function and role in brain disorders. Mol Brain. 2017; 10: 43. [CrossRef]
- Ohlsson R, Paldi A, Graves JAM. Did genomic imprinting and X chromosome inactivation arise from stochastic expression? Trends Genet. 2001; 17: 136-141. [CrossRef]

